Effect of Human XRCC1 Protein Oxidation on the Functional Activity of Its Complexes with the Key Enzymes of DNA Base Excision Repair
I. A. Vasil’eva1#, N. A. Moor1#, and O. I. Lavrik1,2,a*
1Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia2Novosibirsk State University, 630090 Novosibirsk, Russia
# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received November 29, 2019; Revised January 20, 2020; Accepted January 27, 2020
Base excision repair (BER) ensures correction of most abundant DNA lesions in mammals. The efficiency of this multistep DNA repair process that can occur via different pathways depends on the coordinated action of enzymes catalyzing its individual steps. The scaffold XRCC1 (X-ray repair cross-complementing protein 1) protein plays an important coordinating role in the repair of damaged bases and apurinic/apyrimidinic (AP) sites via short-patch (SP) BER pathway, as well as in the repair of single-strand DNA breaks. In this study, we demonstrated for the first time in vitro formation of the ternary XRCC1 complex with the key enzymes of SP BER – DNA polymerase β (Polβ) and DNA ligase IIIα (LigIIIα) – using the fluorescence-based technique. It was found that Polβ directly interacts with LigIIIα, but their complex is less stable than the XRCC1–Polβ and XRCC1–LigIIIα complexes. The effect of XRCC1 oxidation and composition of the multiprotein complex on the efficiency of DNA synthesis and DNA ligation during DNA repair has been explored. We found that formation of the disulfide bond between Cys12 and Cys20 residues as a result of XRCC1 oxidation (previously shown to modulate the protein affinity for Polβ), affects the yield of the final product of SP BER and of non-ligated DNA intermediates (substrates of long-patch BER). The effect of XRCC1 oxidation on the final product yield depended on the presence of AP endonuclease 1. Together with the data from our previous work, the results of this study suggest an important role of XRCC1 oxidation in the fine regulation of formation of BER complexes and their functional activity.
KEY WORDS: DNA base excision repair, protein–protein interactions, human XRCC1 protein, DNA polymerase β, DNA ligase IIIαDOI: 10.1134/S0006297920030049
Abbreviations: APE1, AP endonuclease 1; AP site, apurinic/apyrimidinic site; dRp, deoxyribose phosphate; DTT, dithiothreitol; FAM, 5(6)-carboxyfluorescein; FEN1, flap endonuclease 1; gap-DNA, DNA duplex with one-nucleotide gap; LigI/LigIIIα, DNA ligase I/IIIα; LP BER, long-patch base excision repair; nick-DNA, DNA duplex with single-strand break; PARP1, poly(ADP-ribose) polymerase 1; Polβ/Polδ/Polε, DNA polymerase β/δ/ε; SP BER, short-patch base excision repair; XRCC1, X-ray repair cross-complementing protein 1; XRCC1ox, oxidized form of XRCC1 protein.
Base excision repair (BER) ensures correction of the most abundant DNA
lesions in mammals, such as modified nitrogenous bases,
apurinic/apyrimidinic (AP) sites, and single-strand breaks [1, 2]. BER is one of the essential
systems for genome maintenance. DNA lesion repair via various BER
pathways is presented in Fig. 1. The removal of
damaged bases is initiated by DNA glycosylases specific to particular
types of lesions. Mono- or bifunctional DNA glycosylases catalyze
generation of intact or cleaved (through the β- or
β/δ-elimination mechanism) AP site. Intact AP site is
hydrolyzed by AP endonuclease 1 (APE1); next, DNA polymerase β
(Polβ) removes the deoxyribose phosphate residue (dRp) at the
5′-end of the break due to its dRp lyase activity. The products
formed by the activity of bifunctional DNA glycosylases contain
3′- or 5′-terminal blocking groups which are removed by the
phosphatase activity of polynucleotide kinase/phosphatase (PNKP) or
3′-phosphatase and 3′-phosphodiesterase activities of APE1.
The major pathway of DNA base repair referred to as the short-patch
(SP) BER includes filling the one-nucleotide gap via the DNA polymerase
activity of Polβ and restoration of the strand integrity by DNA
ligase IIIα (LigIIIα). Another pathway, known as the
long-patch (LP) BER, takes place if the 5′-dRp residue is
modified (and cannot be removed by the lyase activity of Polβ).
Strand displacement DNA synthesis is initiated by Polβ and
continued by the replicative DNA polymerases δ and ε
(Polδ and Polε, respectively); the overhanging flap is
removed by flap endonuclease 1 (FEN1), and the final ligation of the
break is catalyzed by DNA ligase I (LigI). The repair of single-strand
DNA breaks originating under the action of damaging factors or during
repair of modified bases and AP sites, involves detection of the break,
removal of the blocking groups, filling the gap, and ligation of the
break. The breaks in DNA are detected mainly by poly(ADP-ribose)
polymerase 1 (PARP1); the 3′- and 5′-ends are deblocked by
APE1, PNKP, aprataxin (APTX), and tyrosyl-DNA phosphodiesterase 1
(TDP1); Polβ and LigIIIα fill the gap and catalyze ligation,
respectively. When interacting with damaged DNA, PARP1 becomes
activated and catalyzes synthesis of poly(ADP-ribose) (PAR) and its
attachment to the enzyme itself and to other proteins participating in
the DNA repair. The major target of this modification (PARylation) in
the BER process is XRCC1 protein (X-ray repair cross-complementing
protein 1) which is recruited to single-strand breaks in chromosomal
DNA mainly by PARP1 [1, 3].
XRCC1 does not exhibit enzymatic activity but acts as a scaffold for
organizing the BER complex [3-5]. XRCC1 contains binding sites for virtually all
proteins participating in BER; these sites are formed by all structural
fragments of this protein, such as the N-terminal (NTD), central
(BRCTa), and C-terminal (BRCTb) domains, and unordered linkers
XL1 and XL2 [4, 5].
Fig. 1. Schematic representation of repair pathways for damaged bases and single-strand DNA breaks. Terminal blocking groups of the DNA breaks are as follows: PUA, 3′-phospho-α,β-unsaturated aldehyde; p, 3′-/5′-phosphate; OH, 3′-/5′-OН-group; dRP, 5′-deoxyribose phosphate; PG, 3′-phosphoglycolate; Ade, 5′-aldehyde group (reproduced from [5] with publisher’s permission).
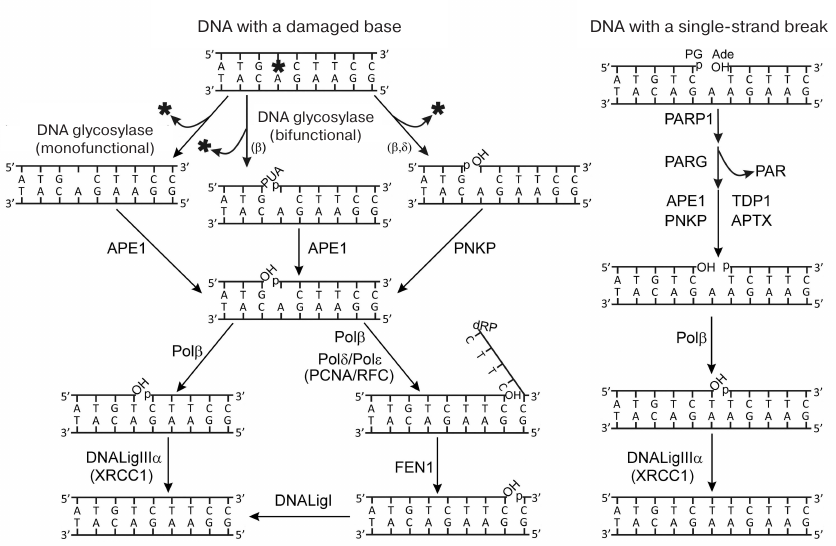
The efficiency of DNA repair in the multistep BER process depends on coordinated activities of enzymes catalyzing its individual steps. One of the coordination mechanisms suggests formation of multiprotein complexes, repairsomes, composed of enzymes and scaffold proteins (e.g., XRCC1) [1, 6]. It has been shown earlier that the XRCC1 complex with LigIIIα is critically important for stabilization of this enzyme and successful DNA repair in vivo [7, 8]. This complex was detected in the cell extracts by non-equilibrium gel filtration and found to be the most stable among protein complexes involving XRCC1 [9]. Using both equilibrium and non-equilibrium methods, we have detected the XRCC1 complex with Polβ and evaluated its stability in comparison with complexes of other proteins [10]. The necessity of this complex for the efficient in vitro and in vivo DNA repair and protein protection against proteasomal degradation was established in [11-13]. Taking in consideration that the binding sites for Polβ and LigIIIα are formed by different XRCC1 domains (NTD and BRCTb, respectively) and do not overlap with the binding sites for other BER participants, we suggested that the ternary complex of these proteins should exist as a stable “platform” for formation of more dynamic multiprotein ensembles [10]. To confirm this hypothesis, we studied LigIIIα interaction with XRCC1 and Polβ by the fluorescent titration method (earlier used by us for characterization of XRCC1 complex with Polβ [10]) in order to compare the affinity of different proteins for each other under identical conditions. The equilibrium method has allowed us to detect complexes of different proteins involved in BER, which could not be detected by the frequently used non-equilibrium methods of affinity co-precipitation and gel-filtration [5, 10]. We also studied the role of ternary complexes containing reduced or oxidized XRCC1 in the regulation of SP BER efficiency in vitro. The disulfide bond formation between the Cys12 and Cys20 residues as a result of XRCC1 oxidation induces structural reorganization of the NTD and its complex with Polβ, as it has been shown by X-ray crystallographic analysis of the complex of these protein domains, and increased the affinity of these proteins for each other [4]. It was found that abolishing the formation of the XRCC1 oxidized form (XRCC1ox) in mammalian cells by replacement of the wild-type XRCC1 with the Cys12Ala mutant reduced the cell survival under oxidative stress [14, 15]. The authors of these studies assumed that formation of a more stable XRCC1ox complex with Polβ is necessary for the efficient in vivo repair of oxidative DNA lesions. However, the effect of XRCC1 oxidation on the DNA synthesis in the process of DNA repair has not been studied.
MATERIALS AND METHODS
Materials. The following reagents were used: [γ-32P]ATP (specific radioactivity, 5 Ci/µmol) from the Laboratory of Biotechnology, Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences; 5(6)-carboxyfluorescein N-hydroxysuccinimide ester (FAM-SE), tris(2-carboxyethyl)phosphine (TCEP) from Sigma (USA); oxidized glutathione (GSSG) from ICN Biomedicals (USA). Reagents for electrophoresis and components of buffer solutions were from Sigma or of domestic production (especially high purity grade). Recombinant mammalian BER proteins were used in the study. Plasmid vectors for expression in Escherichia coli cells of human APE1 and rat Polβ were kindly provided by Dr. Wilson (National Institutes of Health, North Carolina, USA), human XRCC1 – by Dr. Radicella (UMR217 CNRS/CEA, France), and human LigIIIα – by Dr. Dianov (Oxford University, Great Britain). Polβ, XRCC1, and APE1 were prepared as described earlier [16-18]. LigIIIα was prepared using the original method described below. Final preparations of the studied proteins were dialyzed against the storage buffer containing 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 0.5 mM EDTA, 5 mM dithiothreitol (DTT), 40% (v/v) glycerol, and stored at –30°C. DNA oligonucleotides (template, 5′-GGAAGACCCTGACGTTACCCAACTTAATCGCC-3′; primer 1, 5′-GGCGATTAAGTTGGG-3′; primer 2, 5′-GGCGATTAAGTTGGGT-3′; and downstream primer, 5′-pAACGTCAGGGTCTTCC-3′) were synthesized in the Laboratory of Medical Chemistry, Institute of Chemical Biology and Fundamental Medicine. The oligonucleotides were labeled with 32P at the 5′-end using [γ-32P]ATP and T4 polynucleotide kinase (Biosan, Russia) as recommended by the manufacturer. 32-nt DNA duplexes with single-strand breaks (nick-DNA) or one-nucleotide gap (gap-DNA) in the middle of the strand were prepared as described earlier [10].
Preparation of oxidized XRCC1 protein. XRCC1ox was prepared by mild oxidation of SH-groups [19]. XRCC1 (100 μM) was incubated with GSSG (10 mM) in 50 mM Na-phosphate buffer (pH 7.8) containing 100 mM NaCl for 18 h in the dark at 4°C and then dialyzed against DTT-free storage buffer. To prepare fully reduced XRCC1, the original protein preparation was incubated with 10 mM TCEP for 18 h at 4°C and dialyzed against the storage buffer. The content of SH-groups in the XRCC1 and XRCC1ox preparations was determined colorimetrically using the Ellman’s reagent [20].
Expression and purification of DNA ligase IIIα. The full-length human nuclear LigIIIα with the polyhistidine tag at the N-terminus was expressed in E. coli BL21(DE3) Rosetta 2 cells. After transformation with the plasmid, the cells were grown at 30°C in LB medium until A600 0.6; protein synthesis was induced with 1 mM isopropyl-β-D-1-thiogalactopyranoside, and the cells were incubated for another 4 h at 18°C. Next, the cells were precipitated by centrifugation at 7000 rpm at 4°C (Beckman Coulter Inc, USA) and stored at –40°C. The cell biomass was thawed on ice and resuspended in cold lysis buffer containing 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 1% (v/v) NP-40, 1 mM β-mercaptoethanol (b-ME), 10% (v/v) glycerol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine (Bz), and a tablet of inhibitor cocktail (Roche, Switzerland). After incubation with lysozyme (1.5 mg/ml; Boehringer Mannheim, Germany) for 20 min on ice, the cells were disintegrated by sonication (Bandelin, Germany). The resulting suspension was centrifuged (18,000 rpm; Beckman Coulter Inc) at 4°C to remove the cell debris. The supernatant was supplemented with imidazole to the final concentration of 5 mM (pH 8.0) and loaded on Ni-Sepharose 6 Fast Flow (GE Healthcare, Switzerland) equilibrated with buffer A (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 3 mM β-ME, 1 mM PMSF, 1 mM Bz, and 5 mM imidazole). The adsorbed proteins were eluted with 5-600 mM imidazole gradient in buffer A. LigIIIα was purified from protein admixtures by successive chromatographies on Heparin-Sepharose 6 Fast Flow (GE Healthcare) and ssDNA-cellulose (USB, USA) equilibrated with buffer B (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 10 mM β-ME, 0.5 mM EDTA, 5% (v/v) glycerol) using a 0.1-1.0 M gradient of NaCl concentration in buffer B. The obtained fractions were analyzed by electrophoresis in 10% (w/v) SDS-polyacrylamide gel; fractions containing the purified protein were concentrated and dialyzed against the storage buffer in a 10-kDa microdialysis cell (Sartorius, Germany). The activity of LigIIIα was tested in the ligation reaction using synthetic 32-nt duplex with a single-strand break (nick-DNA). The reaction mixture (10 μl) containing 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 10 mM MgCl2, 7 mM DTT, 1 mM ATP, 50 nM 5′-32P-labeled DNA substrate, and 100-400 nM LigIIIα was incubated for 30 min at 37°C. The reaction was initiated by adding the enzyme and terminated by adding an equal volume of the sample buffer [90% formamide (v/v), 20 mM EDTA, 0.05% Bromophenol Blue, and 0.05% Xylene Cyanol]. The samples were incubated for 2 min at 90°C and separated by electrophoresis in 20% polyacrylamide gel with urea. The gels were visualized, and the reaction products were analyzed quantitatively with a Typhoon FLA 9500 scanner (GE Healthcare) and the Quantity One program (Bio-Rad, USA).
Fluorescent labeling of DNA ligase IIIα and investigation of the protein complex formation by the fluorescent titration method. LigIIIα was labeled with FAM-SE at the N-terminal amino group using the method developed by us earlier [10]. The protein was dialyzed against four changes of buffer containing 100 mM MES (pH 7.0) and 200 mM NaCl using a 10-kDa microdialysis cell. The reaction with FAM-SE was performed in the same buffer by incubating 20-50 μM LigIIIα with 40-200 μM reagent for 18 h at 4°C in the dark. The reaction was stopped by adding to the reaction mixture (30-50 μl) the buffer containing 100 mM Hepes (pH 8.0), 200 mM NaCl, and 10 mM DTT to the final volume of 500 μl. The protein was separated from the excessive reagent by gel-filtration on a Spin Trap G-25 microcolumn (GE Healthcare). After concentrating, protein solution was supplemented with glycerol to 40% for storage at –30°C and 0.001% Brij 58 polyoxyethylene cetyl ether (Sigma, USA) to prevent protein aggregation. Protein concentration and the degree of its labeling were determined spectrophotometrically with a CLARIOstar device (GMB Labtech GmbH, Germany) using extinction coefficients ε494 80,445 M–1·cm–1 for FAM and ε280 79,300 M–1·cm–1 for LigIIIα. The specific activity of the labeled enzyme was tested in the ligation reaction using 32P-labeled nick-DNA as described above.
Experiments on the FAM-LigIIIα binding by other proteins were performed by titration of the labeled protein (used at the fixed concentration of 40 nM) either separately with Polβ and XRCC1 (in the absence or presence of the second partner in the concentration of 200 nM) or with an equimolar mixture of XRCC1 and Polβ. The binding buffer contained 50 mM Hepes (pН 8.0), 100 mM NaCl, and 5 mM DTT. The fluorescence intensity of the samples (volume, 12 μl) was measured in non-transparent polypropylene plates (Corning, USA) using a microplate CLARIOstar fluorimeter (GMB Labtech GmbH). The fluorescence excitation wavelength was 485 nm; changes in the fluorescence intensity were detected at the emission wavelength of 530 nm. In each independent experiment, fluorescence intensity was measured at each concentration of the protein partner in three repeats. The experimental data were processed quantitatively with the MARS Data analysis program (GMB Labtech GmbH). The binding curves were described by the equation:
F = F0 + (F∞ – F0)/[1 + (EC50/C)n],
where F0, F, and F∞ are fluorescence intensities of the FAM-LigIIIα solution in the absence of other proteins, in the presence of protein partner in the given (C) and saturating concentrations, respectively; EC50 is the concentration of protein partner at which F – F0 = (F∞ – F0)/2; n is Hill coefficient. All experiments were performed at least three times. The binding curves were plotted using averaged experimental data with the OriginPro 8.6 program; the extent of binding corresponding to the maximal signal at the saturating concentration of the protein partner (F∞ – F0) was taken as a unit, whereas the extent of binding at a given concentration was determined as the ratio (F – F0)/(F∞ – F0).
Registration of the gap-DNA repair products in the presence of enzymes and proteins participating in SP BER. Standard reaction mixture (volume, 10 μl) contained 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 6 mM MgCl2, 50 nM 32P-labeled gap-DNA, and 10-50 nM Polβ. The reaction was initiated by adding a mixture of dATP, dGTP, dTTP, and ddCTP nucleotides (final concentration of each nucleotide, 10 μM), and the reaction mixture was incubated at 37°C for 30 min. When the effect of proteins on the product yield was studied, the reaction mixture contained 50 nM Polβ, 50-200 nM XRCC1 or XRCC1ox, 10-50 nM APE1, and 10-50 nM LigIIIα (in the presence of 1 mM ATP), as indicated in the legends to the figures. The reaction was stopped by adding an equal volume of sample buffer. The samples were incubated for 2 min at 90°C, and the reaction products were separated by electrophoresis in denaturing 20% polyacrylamide gel with urea. The gels were visualized with a Typhoon FLA 9500 scanner and the yield of the reaction products was analyzed with the Quantity One Program.
RESULTS AND DISCUSSION
LigIIIα interaction with XRCC1 and Polβ. Interaction of LigIIIα with the scaffold XRCC1 protein and Polβ catalyzing the synthesis of the LigIIIα substrate in SP BER was studied by the fluorescent titration method. The fluorescence of FAM-labeled LigIIIα was measured in the absence and presence of unlabeled XRCC1 and Polβ. To label LigIIIα with FAM-SE preferentially at the N-terminal amino group, the reaction was performed at pH 7.0 [21]. The reaction conditions were optimized by varying the protein and reagent concentrations in order to obtain fully active protein that would contain no more than one fluorescein residue per protein molecule. Protein preparations with the labeling stoichiometry of ~0.5 mol FAM per 1 mol protein were obtained by incubating 50 μM protein with 200 μM reagent.
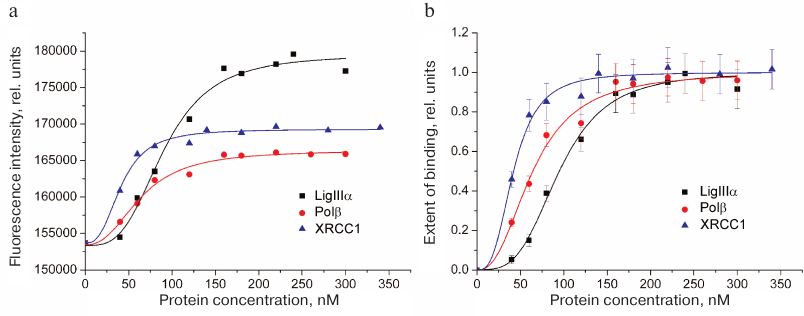
Typical curves of FAM-LigIIIα titration with unlabeled proteins (LigIIIα, Polβ, and XRCC1) and the corresponding protein binding curves are presented in Fig. 2. The maximal increase in the fluorescence intensity of the labeled protein in the presence of saturating concentrations of protein partners did not exceed 20% (in contrast to earlier recorded 1.5- to 2.0-fold fluorescence intensity changes for other FAM-labeled proteins) [10]. This difference can be explained by a considerable distance between the fluorophore in FAM-LigIIIα and C-terminal BRCT domain containing the sites for both LigIIIα homo-association and protein hetero-association with XRCC1 [5]. The effective concentration of the protein partner (EC50) at which the increase in the fluorescence intensity over the initial level (F – F0) amounts to a half of the maximal increase at the saturating concentration [(F∞ – F0)/2] is the apparent equilibrium constant of complex dissociation. Comparison of this parameter for different proteins (table) suggests formation of more stable complexes upon hetero-association than upon homo-association of LigIIIα. The affinity of LigIIIα for XRCC1 (EC50 = 42 nM) is 1.5 times higher than for Polβ (EC50 = 64 nM). However, XRCC1 has a higher affinity for Polβ (EC50 = 23 nM [10]) than for LigIIIα. In general, XRCC1 complexes with Polβ and LigIIIα are the most stable among all complexes of proteins participating in BER that had been characterized by us using the same equilibrium method [10].
Quantitative characteristics of LigIIIα interaction with BER
proteins
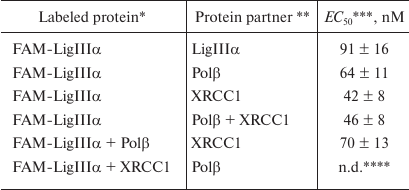
* FAM-LigIIIα (40 nM) was titrated with the protein partner in the
absence or presence of Polβ or XRCC1 pre-added at excessive
concentrations (200 nM).
** FAM-LigIIIα was titrated with individual proteins or
with an equimolar mixture of Polβ and XRCC1.
*** Apparent equilibrium dissociation constants of protein complexes
are shown as mean ± standard deviation from 3-5 independent
experiments.
**** Not determined because of the absence of changes in the
fluorescence intensity.
Fig. 2. Homo- and hetero-association of LigIIIα with proteins participating in BER as studied by fluorescent titration. a) Typical curves for FAM-labeled LigIIIα (40 nM) titration with unlabeled proteins (LigIIIα, Polβ, and XRCC1); b) binding curves calculated from the data of 3-5 independent experiments (mean ± standard deviation).
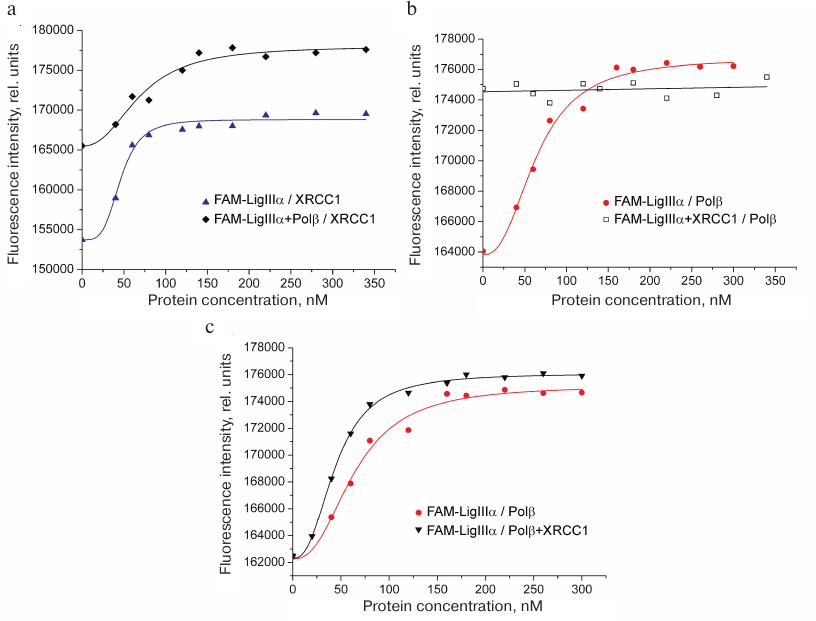
To detect the formation of the ternary XRCC1 complex with LigIIIα and Polβ, increasing concentrations of XRCC1 or Polβ were added to the FAM-LigIIIα mixture containing either Polβ or XRCC1, respectively, at high (sub-saturating) concentrations. In each experiment, FAM-LigIIIα was titrated with the corresponding protein, and the fluorescence of the protein mixture was recorded. Higher background fluorescence (F0) for the FAM-LigIIIα mixtures with Polβ or XRCC1 in comparison with FAM-LigIIIα (Fig. 3, a and b) was caused by the formation of the binary complex. The increase in fluorescence upon XRCC1 addition to the FAM-LigIIIα mixture with Polβ (Fig. 3a) suggested XRCC1 interaction with FAM-LigIIIα, despite the presence of excessive concentrations of more preferable XRCC1 interaction partner. The EC50 value for the FAM-LigIIIα binding to XRCC1 in the ternary mixture (70 nM) was lower than the total XRCC1 concentration necessary for independent half-binding of FAM-LigIIIα (42 nM) and half-binding of Polβ (no less than 100 nM), which indicated the ternary complex formation. The absence of changes in the fluorescence signal of the FAM-LigIIIα mixture with XRCC1 after addition of Polβ (Fig. 3b) indicated that Polβ binding to XRCC1 resulting in the ternary complex formation did not noticeably influence the interaction between XRCC1 and FAM-LigIIIα. Titration of FAM-LigIIIα with the equimolar mixture of Polβ and XRCC1 caused a higher increase in the fluorescence intensity than titration with Polβ alone (Fig. 3c). This suggests different environment of the fluorophore upon formation of the FAM-LigIIIα binary complex with Polβ and ternary complex with Polβ and XRCC1. The EC50 value for the FAM-LigIIIα binding in the ternary mixture (46 nM) was comparable with the EC50 value for its binary complex with XRCC1 (42 nM). Taken together, these data evidence preferential interaction of LigIIIα with XRCC1 in the ternary complex with Polβ. Hence, we demonstrated for first time the direct interaction between the Polβ and LigIIIα enzymes catalyzing successive steps of SP BER, as well as formation of their ternary complex with the scaffold protein XRCC1 (Polβ•XRCC1•LigIIIα). Direct interaction of Polβ with the LigI enzyme completing LP BER was shown earlier [22].
Fig. 3. Formation of the ternary complex between LigIIIα, Polβ, and XRCC1. Typical fluorescence curves of FAM-LigIIIα (40 nM) titration with (a) XRCC1 in the absence and presence of Polβ (200 nM), (b) Polβ in the absence and presence of XRCC1 (200 nM), and (c) Polβ or equimolar mixture of Polβ and XRCC1. The binding curves calculated from the averaged data of independent experiments are presented in Fig. S1 in the Supplement to this paper on the web site of the journal (http://protein.bio.msu.ru/biokhimiya) and Springer site (Link.springer.com).
Effect of other proteins on DNA synthesis by Polβ. To study the influence of protein complexes of different composition on the activity of Polβ in the DNA repair synthesis, we preliminary determined concentrations of individual proteins at which Polβ was activated (Fig. 4). Both Polβ concentration and incubation time were optimized to test the products of the SP and LP DNA synthesis (filling the gap in gap-DNA and strand displacement DNA synthesis, respectively) upon incomplete conversion of the substrate. To limit the length of the synthesized products in the case of strand displacement DNA synthesis, the reaction mixture contained a dideoxynucleotide (ddCTP). The stimulatory effect of the reduced XRCC1 on strand displacement DNA synthesis increased with the increase in the protein concentration from 50 to 200 nM: the fraction of the product corresponding to the insertion of one nucleotide into the repaired strand (S+1) decreased, whereas the fraction of the product corresponding to the insertion of four nucleotides (S+4) increased (Fig. 4, lanes 6-8). In the case of oxidized XRCC1 (XRCC1ox), the effect did not depend on the protein concentration (50-200 nM) (Fig. 4, lanes 9-11). For comparison, in subsequent experiments (in the presence of other proteins), we used XRCC1 at a concentration of 100 nM, which allowed us to observe the effect of this protein on the efficiency of the initial substrate conversion, as well as the difference in the effects produced by the reduced and oxidized XRCC1. Beside the catalytic conversion of the product of gap filling (S+1) into the final product of the SP BER, LigIIIα stimulated strand displacement DNA synthesis; this effect decreased with the increase in the enzyme concentration due to more efficient ligation (Fig. 4, lanes 12-14). The concentration of 50 nM was chosen, at which the fraction of the initial substrate decreased noticeably. The stimulatory effect of APE1 on the strand displacement DNA synthesis and efficiency of the initial substrate conversion decreased with the increase in the protein concentration from 10 to 50 nM because of the 3′-5′-exonuclease activity of the enzyme (Fig. 4, lanes 3-5); however, it could still be observed at the concentration of 50 nM used in subsequent experiments.
Fig. 4. Effect of different concentrations of BER proteins on the Polβ activity in the DNA repair synthesis. Gel autoradiograph of the products of the Polβ-catalyzed DNA synthesis in the absence and presence of other proteins is shown; the histogram demonstrates relative yields of products of DNA strand elongation and ligation in the corresponding samples. 32P-labeled gap-DNA (50 nM) was incubated with Polβ (50 nM) and a mixture of dATP, dGTP, dTTP, and ddCTP (10 μM of each nucleotide) for 30 min at 37°C (lanes 2-14) in the absence or presence of APE1 (10-50 nM), XRCC1 (50-200 nM), XRCC1ox (50-200 nM), or LigIIIα (10-50 nM) (see “Materials and Methods” for the detailed reaction conditions). S, 32P-labeled primer (15-nt) in the initial DNA; S+1 to S+4, products of primer elongation; P, final product of DNA repair after ligation (32-nt); the corresponding structures of duplexes (intermediates at different steps of SP/LP BER) are presented in the scheme to the right; asterisk shows position of the 32P-label. The total yield of the products with one or two inserted dAMP residues (S+2 and S+3) is shown, because these products could not be resolved in the gel. The data of one of three independent experiments are presented; deviation of the relative yield values from the mean values was 2-10%.
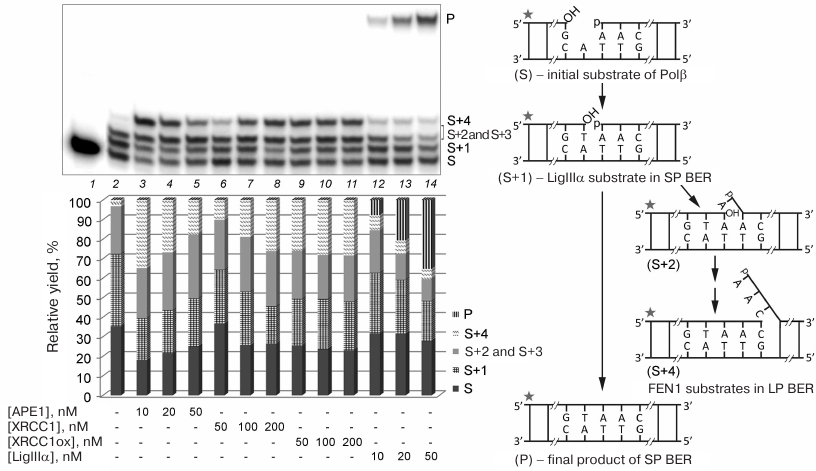
The data presented in Fig. 5 show that APE1, XRCC1, XRCC1ox, and LigIIIα produced comparable effects on the total efficiency of initial substrate conversion; the content of the substrate decreased ~10% in all the cases (lanes 3, 4, 5, and 8 vs. lane 2). According to their stimulatory effect on the strand displacement DNA synthesis (increase in the yield of S+4), the proteins can be arranged in the following order: XRCC1ox > XRCC1 > APE1 > LigIIIα. The stimulatory effect of XRCC1ox significantly exceeded that of XRCC1: the S+4 fraction in the presence of XRCC1ox was two times greater than in the presence of XRCC1, which might be explained by a higher affinity of XRCC1ox for Polβ [4].
Fig. 5. Effect of BER proteins on the DNA synthesis by Polβ. Autoradiograph of the gel with the products of the DNA repair synthesis catalyzed by Polβ in the absence and presence of APE1, XRCC1, XRCC1ox, or LigIIIα is presented. The histogram shows the relative yields of the DNA strand elongation/ligation reaction. 32P-labeled gap-DNA (50 nM) was incubated with Polβ (50 nM) and a mixture of dATP, dGTP, dTTP, and ddCTP (10 μM of each nucleotide) for 30 min at 37°C (lanes 2-13) in the absence or presence (+) of APE1 (50 nM), different forms of XRCC1 (100 nM), or LigIIIα (50 nM) (see “Materials and Methods” for the detailed reaction conditions). The total yield of the products with one or two inserted dAMP residues (S+2 and S+3) is shown, because these products could not be resolved in the gel. For designations, see the legend to Fig. 4. The data of one of three independent experiments are presented; deviation of the relative yield values from the mean values was 2-10%.
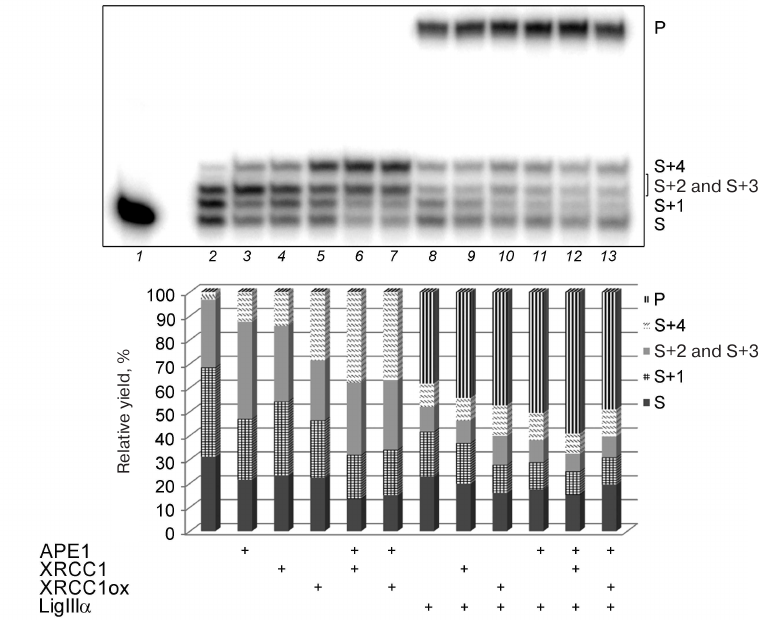
APE1 enhanced the stimulatory effect of XRCC1 on the strand displacement DNA synthesis to a greater extent than the stimulatory effect of XRCC1ox: the S+4 fraction increased 2.7-fold in the first case vs. 1.3-fold in the second case (Fig. 5, lanes 6 and 7 vs. lanes 4 and 5). As a result, the difference between the effects of reduced and oxidized XRCC1 in the presence of APE1 was eliminated. Formation of the XRCC1 ternary complex with Polβ and APE1 was shown by us earlier [10]. The binding site for APE1 is located in the linker region (XL1) adjacent to the XRCC1 N-terminal domain (NTD) containing the binding site for Polβ [4]. It cannot be excluded that XRCC1 oxidation induces rearrangement not only of the NTD, where the disulfide bond is formed between the Cys12 and Cys20 residues, but also of the whole three-dimensional protein structure. More stable binding of XRCC1ox to Polβ can have a negative effect on the formation of the ternary complex with APE1.
The combined effect of LigIIIα and XRCC1ox on the efficiency of substrate conversion is greater than the combined effect of LigIIIα and XRCC1 (Fig. 5, lane 10 vs. lane 9). Although LigIIIα suppressed the stimulatory effect of both XRCC1 forms on strain displacement DNA synthesis, XRCC1ox was still able to stimulate this synthesis more efficiently than XRCC1 even in the presence of LigIIIα (lanes 9 and 10 vs. lanes 4 and 5 and vs. each other). Both XRCC1 and XRCC1ox promoted the activity of LigIIIα: the yield of the final product of DNA repair increased by 6 and 9% in the presence of XRCC1 and XRCC1ox, respectively (lanes 9 and 10 vs. lane 8). Interestingly, the total yield of the ligated intermediate (S+1) and the final product (P) of SP BER was similar in the combined presence of LigIIIα and XRCC1 proteins (61%) and LigIIIα and XRCC1ox proteins (59%), but the degree of S+1 conversion into the final product was higher in the second case (72 vs. 80%, respectively). It may be assumed that the ternary complex formation and stabilization of the Polβ interaction with XRCC1ox (in comparison with the Polβ complex with XRCC1) promotes the transfer of DNA intermediate from one enzyme to the other.
The efficiency of substrate conversion in the combined presence of APE1 and LigIIIα exceeded that in the presence of individual proteins (Fig. 5, lane 11 vs. lanes 3 and 8). APE1 stimulated LigIIIα activity more efficiently than XRCC1 and XRCC1ox and increased the yield of the final product by 12% (lane 11 vs. lanes 9 and 10). LigIIIα suppressed the stimulatory effect of APE1 on the synthesis of non-ligated intermediates (S+2, S+3, and S+4): the yield of these products in the presence of APE1 and LigIIIα was 2.5 times lower than in the presence of APE1 and nearly the same as in the presence of LigIIIα (lane 11 vs. lanes 3 and 8). The activating effect of APE1 on DNA ligation was also observed for LigI, which indicates an active role of the enzyme responsible for deblocking the 3′-ends in the breaks in coordination of the entire process of DNA repair [23].
When the reaction mixture contained all proteins participating in SP BER, the highest DNA repair efficiency was observed for the complexes containing XRCC1: the yield of final product in this case was 10% higher than in the presence of XRCC1ox (Fig. 5, lane 12 vs. lane 13). Moreover, the efficiency of DNA repair in the combined presence of APE1, LigIIIα, and XRCC1 was higher than in the presence of two proteins: APE1 and LigIIIα (by 9%) or LigIIIα and XRCC1 (by 15%) (lane 12 vs. lanes 11 and 9, respectively). This can be caused by a combination of the stronger effect of APE1 vs. XRCC1 (or XRCC1ox) on the activity of LigIIIα and the lower stimulatory effect of XRCC1 vs. XRCC1ox on the synthesis of non-ligated DNA intermediates, which might indicate formation of more stable multiprotein ensemble with the participation of all the proteins in the XRCC1 case. Indeed, the distribution of products in the presence of LigIIIα and XRCC1ox changed insignificantly upon addition of APE1 (lane 10 vs. lane 13). Therefore, the influence of XRCC1 oxidation on the efficiency of DNA repair catalyzed by Polβ via the SP pathway depends on the repairosome composition.
Using equilibrium fluorescent titration, we have shown for the first time in vitro formation of the ternary complex by the major participants of SP BER: enzymes Polβ and LigIIIα and the scaffold protein XRCC1. Polβ and LigIIIα, whose direct interaction in the absence of XRCC1 was also demonstrated for the first time, form a complex which is less stable than the Polβ–XRCC1 and LigIIIα–XRCC1 complexes. This might explain why the ternary complex formation provides more efficient coordination of the BER final steps. No ternary complex has been detected in the cell extracts [9], probably, because of its instability under non-equilibrium conditions of gel-filtration and/or destabilizing action of other proteins in the extract. However, in another work, XRCC1, Polβ, and LigIIIα were found to be present together in many multiprotein BER complexes isolated from the nuclear extracts [24]. The existence of the ternary complex in vivo was confirmed by synchronous localization of XRCC1, Polβ, and LigIIIα on DNA lesions [25].
The stability of the XRCC1 complex with Polβ is regulated by oxidation of the former protein resulting in the disulfide bond formation between Cys12 and Cys20 residues in the N-terminal domain containing the binding site for Polβ [4]. XRCC1ox is present in mammalian cells; the relative content of this XRCC1 form determines the cytotoxicity of oxidative agents caused by BER deficiency [14, 15]. A pronounced difference between the survival of cells expressing wild-type XRCC1 and its Cys12Ala mutant (incapable of oxidation) has been revealed only under extreme oxidative stress conditions caused by suppression of glutathione synthesis [15]. It was shown that the presence of XRCC1ox accelerated Polβ accumulation on DNA lesions [14, 15]; however, it was still unclear how the oxidative modification of XRCC1 influences formation of DNA repair complexes and the process of DNA repair itself. Using the reconstituted system, we showed that formation of the ternary complex and XRCC1 oxidation determine the efficiency of DNA repair via the SP BER. The stimulatory effect of XRCC1ox on the catalytic activity of Polβ and LigIIIα was more pronounced than that of the reduced XRCC1. However, in the presence of APE1, XRCC1 oxidation decreased the yield of the final product of DNA repair. Therefore, stimulation of the DNA synthesis by XRCC1 oxidation can be important for the efficient recovery of some DNA lesions. In particular, the repair of some single-strand breaks and damaged bases removed by bifunctional DNA glycosylases by the β/δ elimination mechanism does not require APE1 (Fig. 1).
The composition of the BER protein complexes depends on the type and level of DNA lesions, and the repair of extensive lesions needs participation of PARP1 for more efficient assembly of the DNA repair complexes [24]. The level of PARP1 automodification by the synthesized ADP-ribose polymer (PAR) influences the PARP1 affinity for DNA intermediates of BER and is an important factor in the DNA repair regulation [1, 3]. XRCC1 possesses a high affinity for PAR and plays a paramount role in the assembly of the DNA repair complexes involving PARP1 [26]. Moreover, XRCC1 is a target of ADP-ribosylation and regulates the level of PARP1 automodification by inhibiting PAR elongation [27]. As we have established recently, XRCC1 oxidation has a negative effect on the interaction of this protein with PAR and its acceptor activity in the reaction of ADP-ribosylation, which suppresses the inhibitory effect of the protein on PARP1 automodification [28]. The difference between the reduced and oxidized forms of XRCC1 in their ability to regulate the level of PARP1 automodification increases in the presence of Polβ [28], suggesting a cooperation between XRCC1 and Polβ in this process. Hence, the ratio between XRCC1 and XRCC1ox content can regulate the lifetime of the PAR–PARP1 complex with the damaged DNA. We found in this study that the presence of XRCC1ox instead of XRCC1 in the majority of studied complexes increased the fraction of non-ligated DNA intermediates, thereby requiring more efficient DNA repair in the repeated cycle. These lesions can be repaired via the LP BER with the involvement of Polβ and FEN1. In its turn, the activity of Polβ in LP BER is regulated by PARP1 and its PARylation: PARP1 suppresses DNA synthesis and FEN1-dependent stimulation of DNA repair, but its inhibitory effect decreases upon PARP1 automodification [29]. At the same time, PARP1 interaction with DNA intermediates of SP BER does not influence the efficiency of DNA repair via this pathway because of the absence of PARP1 inhibitory action on the dRp lyase activity of Polβ and ligase activity of LigIIIα [29-31]. Therefore, by ensuring fine regulation of PARP1 automodification, XRCC1 oxidation can provide more efficient repair of non-ligated DNA intermediates in the repeated cycle with the participation of PARP1. Based on the results of our previous work [28] and this study, we can conclude that the redox status of XRCC1 influences the efficiency of BER both at the stage of DNA repair complex formation (via regulation of PARP1 automodification) and the stage of DNA repair synthesis.
Funding. This work was supported by the Program for Basic Research of the State Academies of Sciences 2013-2020 (project AAAA-A17-117020210022-4; preparation of recombinant proteins) and Russian Science Foundation (project 19-14-00107; study of protein–protein interactions).
Acknowledgements. The authors are grateful to Dr. S. H. Wilson (National Institutes of Health, North Carolina, USA), Dr. J. P. Radicella (UMR217 CNRS/CEA, France), and Dr. G. Dianov (Oxford University, UK) for kindly providing plasmid vectors for protein expression and to the student Zhao Mingxing of the Novosibirsk State University for participating in the fluorescent titration experiments .
Conflict of interest. The authors declare no conflict of interest.
Compliance with ethical norms. The present article does not contain description of studies involving human or animal subjects performed by any of the authors.
REFERENCES
1.Abbotts, R., and Wilson, D. M., 3rd (2017)
Coordination of DNA single strand break repair, Free Radic. Biol.
Med., 107, 228-244, doi:
10.1016/j.freeradbiomed.2016.11.039.
2.Whitaker, A. M., Schaich, M. A., Smith, M. R.,
Flynn, T. S., and Freudenthal, B. D. (2017) Base excision repair of
oxidative DNA damage: from mechanism to disease, Front. Biosci.
(Landmark Ed.), 22, 1493-1522, doi: 10.2741/4555.
3.Caldecott, K. W. (2014) DNA single-strand break
repair, Exp. Cell. Res., 329, 2-8, doi:
10.1016/j.yexcr.2014.08.027.
4.London, R. E. (2015) The structural basis of
XRCC1-mediated DNA repair, DNA Repair (Amst.), 30,
90-103, doi: 10.1016/j.dnarep.2015.02.005.
5.Moor, N. A., and Lavrik, O. I. (2018)
Protein–protein interactions in DNA base excision repair,
Biochemistry (Moscow), 83, 411-422, doi:
10.1134/S0006297918040120.
6.Dutta, A., Yang, C., Sengupta, S., Mitra, S., and
Hegde, M. L. (2015) New paradigms in the repair of oxidative damage in
human genome: mechanisms ensuring repair of mutagenic base lesions
during replication and involvement of accessory proteins, Cell. Mol.
Life Sci., 72, 1679-1698, doi:
10.1007/s00018-014-1820-z.
7.Caldecott, K. W., McKeown, C. K.,
Tucker, J. D., Ljungquist, S., and Thompson, L. H. (1994)
An interaction between the mammalian DNA repair protein XRCC1 and DNA
ligase III, Mol. Cell. Biol., 14, 68-76, doi:
10.1128/mcb.14.1.68.
8.Kubota, Y., and Horiuchi, S. (2003)
Independent roles of XRCC1′s two BRCT motifs in recovery from
methylation damage, DNA Repair (Amst.), 2, 407-415, doi:
10.1016/s1568-7864(02)00242-2.
9.Parsons, J. L., Dianova, I. I.,
Allinson, S. L., and Dianov, G. L. (2005) DNA polymerase beta
promotes recruitment of DNA ligase III alpha-XRCC1 to sites of base
excision repair, Biochemistry, 44, 10613-10619, doi:
10.1021/bi050085m.
10.Moor, N. A., Vasil'eva, I. A., Anarbaev, R. O.,
Antson, A. A., and Lavrik, O. I. (2015) Quantitative characterization
of protein–protein complexes involved in base excision DNA
repair, Nucleic Acids Res., 43, 6009-6022, doi:
10.1093/nar/gkv569.
11.Kubota, Y., Nash, R. A., Klungland, A.,
Schär, P., Barnes, D. E., and Lindahl, T. (1996) Reconstitution of
DNA base excision-repair with purified human proteins: interaction
between DNA polymerase β and the XRCC1 protein, EMBO J.,
15, 6662-6670, doi: 10.1002/j.1460-2075.1996.tb01056.x.
12.Wong, H. K., and Wilson, D. M., 3rd (2005)
XRCC1 and DNA polymerase beta interaction contributes to cellular
alkylating agent resistance and single-strand break repair, J. Cell.
Biochem., 95, 794-804, doi: 10.1002/jcb.20448.
13.Fang, Q., Inanc, B., Schamus,
S., Wang, X. H., Wei, L., Brown, A. R.,
Svilar, D., Sugrue, K. F., Goellner, E. M., Zeng,
X., Yates, N. A., Lan, L., Vens, C., and
Sobol, R. W. (2014) HSP90 regulates DNA repair via the interaction
between XRCC1 and DNA polymerase β, Nat. Commun., 5,
5513, doi: 10.1038/ncomms6513.
14.Horton, J. K., Stefanick, D. F., Gassman, N. R.,
Williams, J. G., Gabel, S. A., Cuneo, M. J., Prasad, R., Kedar, P. S.,
Derose, E. F., Hou, E. W., London, R. E., and Wilson, S. H. (2013)
Preventing oxidation of cellular XRCC1 affects PARP-mediated DNA damage
responses, DNA Repair (Amst.), 12, 774-785, doi:
10.1016/j.dnarep.2013.06.004.
15.Horton, J. K., Seddon, H. J., Zhao, M. L.,
Gassman, N. R., Janoshazi, A. K., Stefanick, D. F., and Wilson, S. H.
(2017) Role of the oxidized form of XRCC1 in protection against extreme
oxidative stress, Free Radic. Biol. Med., 107, 292-300,
doi: 10.1016/j.freeradbiomed.2017.02.005.
16.Kumar, A., Widen, S. G., Williams, K. R., Kedar,
P., Karpel, R. L., and Wilson, S. H. (1990) Studies of the domain
structure of mammalian DNA polymerase beta. Identification of a
discrete template binding domain, J. Biol. Chem., 265,
2124-2131.
17.Belousova, E. A., Vasil'eva, I. A., Moor, N. A.,
Zatsepin, T. S., Oretskaya, T. S., and Lavrik, O. I. (2013) Clustered
DNA lesions containing 5-formyluracil and AP site: repair via the BER
system, PLoS One, 8, e68576, doi:
10.1371/journal.pone.0068576.
18.Strauss, P. R., Beard, W. A., Patterson, T. A.,
and Wilson, S. H. (1997) Substrate binding by human
apurinic/apyrimidinic endonuclease indicates a Briggs–Haldane
mechanism, J. Biol. Chem., 272, 1302-1307, doi:
10.1074/jbc.272.2.1302.
19.Chim, N., Harmston, C. A., Guzman, D. J., and
Goulding, C. W. (2013) Structural and biochemical characterization of
the essential DsbA-like disulfide bond forming protein from
Mycobacterium tuberculosis, BMC Struct. Biol., 13,
23, doi: 10.1186/1472-6807-13-23.
20.Wunderlich, M., and Glockshuber, R. (1993)
Redox properties of protein disulfide isomerase (DsbA) from
Escherichia coli, Protein Sci., 2, 717-726, doi:
10.1002/pro.5560020503.
21.Haugland, R. P. (2005) The Handbook – A
Guide to Fluorescent Probes and Labeling Technologies, 10th Edn.,
Invitrogen Corp., USA.
22.Dimitriadis, E. K., Prasad, R., Vaske, M. K.,
Chen, L., Tomkinson, A. E., Lewis, M. S., and Wilson, S. H. (1998)
Thermodynamics of human DNA ligase I trimerization and association with
DNA polymerase beta, J. Biol. Chem., 273, 20540-20550,
doi: 10.1074/jbc.273.32.20540.
23.Ranalli, T. A., Tom, S., and Bambara, R. A.
(2002) AP endonuclease 1 coordinates flap endonuclease 1 and DNA ligase
I activity in long patch base excision repair, J. Biol. Chem.,
277, 41715-41724, doi: 10.1074/jbc.M207207200.
24.Hanssen-Bauer, A., Solvang-Garten, K., Sundheim,
O., Pena-Diaz, J., Andersen, S., Slupphaug, G., Krokan, H. E., Wilson,
D. M., 3rd, Akbari, M., and Otterlei, M. (2011) XRCC1 coordinates
disparate responses and multiprotein repair complexes depending on the
nature and context of the DNA damage, Environ. Mol. Mutagen.,
52, 623-635, doi: 10.1002/em.20663.
25.Lan, L., Nakajima, S., Oohata, Y., Takao, M.,
Okano, S., Masutani, M., Wilson, S. H., and Yasui, A. (2004) In
situ analysis of repair processes for oxidative DNA damage in
mammalian cells, Proc. Natl. Acad. Sci. USA, 101,
13738-13743, doi: 10.1073/pnas.0406048101.
26.Polo, L. M., Xu, Y., Hornyak, P., Garces, F.,
Zeng, Z., Hailstone, R., Matthews, S. J., Caldecott, K. W., Oliver, A.
W., and Pearl, L. H. (2019) Efficient single-strand break repair
requires binding to both poly(ADP-ribose) and DNA by the central BRCT
domain of XRCC1, Cell Rep., 26, 573-581, doi:
10.1016/j.celrep.2018.12.082.
27.Masson, M., Niedergang, C., Schreiber, V., Muller
S., Menissier-de Murcia, J., and de Murcia, G. (1998) XRCC1 is
specifically associated with poly(ADP-ribose) polymerase and negatively
regulates its activity following DNA damage, Mol. Cell. Biol.,
18, 3563-3571, doi: 10.1128/mcb.18.6.3563.
28.Vasil'eva, I. A., Moor, N. A., and Lavrik, O. I.
(2019) Role of oxidation of XRCC1 protein in regulation of mammalian
DNA repair process, Doklady Biochem. Biophys., 489,
357-361, doi: 10.1134/S1607672919060012.
29.Sukhanova, M., Khodyreva, S., and Lavrik, O.
(2010) Poly(ADP-ribose) polymerase 1 regulates activity of DNA
polymerase beta in long patch base excision repair, Mutat. Res.,
685, 80-89, doi: 10.1016/j.mrfmmm.2009.08.009.
30.Prasad, R., Lavrik, O. I., Kim, S. J., Kedar, P.,
Yang, X. P., Vande Berg, B. J., and Wilson, S. H. (2001) DNA polymerase
beta-mediated long patch base excision repair. Poly(ADP-ribose)
polymerase-1 stimulates strand displacement DNA synthesis, J. Biol.
Chem., 276, 32411-32414, doi: 1074/jbc.C100292200.
31.Leppard, J. B., Dong, Z., Mackey, Z. B., and
Tomkinson, A. E. (2003) Physical and functional interaction between DNA
ligase IIIalpha and poly(ADP-ribose) polymerase 1 in DNA single-strand
break repair, Mol. Cell. Biol., 23, 5919-5927, doi:
10.1128/mcb.23.16.5919-5927.2003.
Supplementary Figure S1 (PDF)