Effect of Structure Variations in the Inter-subunit Contact Zone on the Activity and Allosteric Regulation of Inorganic Pyrophosphatase from Mycobacterium tuberculosis
R. S. Romanov1, S. A. Kurilova1, A. A. Baykov1, and E. V. Rodina2,a*
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia2Lomonosov Moscow State University, Faculty of Chemistry, 119991 Moscow, Russia
* To whom correspondence should be addressed.
Received December 26, 2019; Revised January 14, 2020; Accepted January 14, 2020
Hexameric inorganic pyrophosphatase from Mycobacterium tuberculosis (Mt-PPase) has a number of structural and functional features that distinguish it from homologous enzymes widely occurring in living organisms. In particular, it has unusual zones of inter-subunit contacts and lacks the N-terminal region common for other PPases. In this work, we constructed two mutant forms of the enzyme, Ec-Mt-PPase and R14Q-Mt-PPase. In Ec-Mt-PPase, the missing part of the polypeptide chain was compensated with a fragment of PPase from Escherichia coli (Ec-PPase). In R14Q-Mt-PPase, a point mutation was introduced to the contact interface between the two trimers of the hexamer. Both modifications significantly improved the catalytic activity of the enzyme and abolished its inhibition by the cofactor (Mg2+ ion) excess. Activation of Mt-PPase by low (~10 μM) concentrations of ATP, fructose-1-phosphate, L-malate, and non-hydrolyzable substrate analogue methylene bisphosphonate (PCP) was observed. At concentrations of 100 μM and higher, the first three compounds acted as inhibitors. The activating effect of PCP was absent in both mutant forms, and the inhibitory effect of fructose-1-phosphate was absent in Ec-Mt-PPase. The effects of other modulators varied only quantitatively among the mutants. The obtained data indicate the presence of allosteric sites in Mt-PPase, which are located in the zones of inter-subunit contact or associated with them.
KEY WORDS: pyrophosphatase, site-directed mutagenesis, allosteric regulation, fructose-1-phosphate, L-malateDOI: 10.1134/S0006297920030086
Abbreviations: PPase, inorganic pyrophosphatase; Ec-PPase, PPase from Escherichia coli; Mt-PPase, inorganic pyrophosphatase from Mycobacterium tuberculosis; Ec-Mt-PPase, chimeric enzyme in which Mt-PPase polypeptide chain is extended at the N-terminus with residues 1-12 of Ec-PPase; R14Q-Mt-PPase, mutant variant of Mt-PPase with Arg14 replaced by Gln; PCP, methylene bisphosphonate.
Family I soluble inorganic pyrophosphatases (PPases) catalyze hydrolysis
of inorganic pyrophosphate with the formation of orthophosphate. PPases
are present in the majority of living organisms, where they hydrolyze
pyrophosphate, thus shifting multiple processes accompanied by its
formation towards the generation of products [1].
PPases from different organisms have similar active site structure and
mechanism of action and exhibit a high degree of conservation of amino
acid sequences and subunit spatial structure [2].
All PPases are active only in the presence of magnesium ions, which
activate both the enzyme and the substrate. In total, three magnesium
ions per active site are required for catalysis: one of them forms true
substrate with pyrophosphate (MgPPi complex); the other two
form the substrate-binding site in the enzyme and activate nucleophilic
water molecule [3]. The enzymatic reaction proceeds
without formation of a covalent intermediate via direct attack of
phosphorus atom by water molecule activated with two magnesium ions [4].
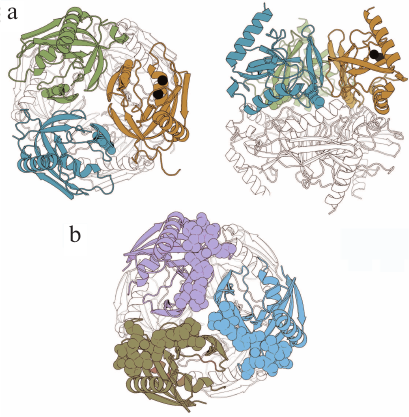
PPase from Mycobacterium tuberculosis (Mt-PPase) exhibits a number of structural and functional features that distinguish it from the majority of known PPases. For this reason, it was included into the group of potential targets for treating animal and human tuberculosis [5]. Hybrid nanomaterials based on immobilized Mt-PPases as active agents show potential in the treatment of human pyrophosphate arthropathy [6]. The enzyme is a hexamer formed by a dimer of trimers (Fig. 1a). The absence of 12 amino acid residues at the N-terminus of the polypeptide chain typical of PPase from Escherichia coli (Ec-PPase) (Fig. 1b) and other representatives of prokaryotic PPases is one of the structural features of this enzyme that affects the inter-subunit contacts. Furthermore, the active site of Mt-PPase contains two histidine residues (at positions 21 and 86) instead of the more common alanine and lysine residues [7].
Fig. 1. Three-dimensional structures of hexameric Mt-PPase and Ec-PPase (PDB accession numbers: 4Z71 and 1OBW, respectively) [8, 9]. a) Two views of Mt-PPase structure – along the third-order rotational symmetry axis (on the left) and perpendicular to the axis (on the right). Subunits of the upper trimer are of different colors; the lower trimer is not colored. The N-terminal residues of each chain are shown in the respective color. The substituted Arg14 residue is presented as a ball-and-stick model in all subunits of the colored trimer. Magnesium ions located in the active site are shown as black spheres. b) View along the third-order rotational symmetry axis of the Ec-PPase molecule. Subunits of the upper trimer are of different colors; lower trimer is not colored. Residues 1-12 absent in Mt-PPase are shown in brighter color in the subunits of the upper trimer. The images are generated with the UCSF Chimera program (UCSF Resource for Biocomputing, Visualization, and Informatics, USA) [10].
To elucidate the pathways of Mt-PPase regulation and to search for the structure-function relationship in the enzyme molecule, we constructed mutant variants of Mt-PPase with the modified inter-subunit contacts and studied the effects of these modifications on the catalytic properties of Mt-PPase and its interactions with the substrate analogue and three potential allosteric regulators. The obtained information on the Mt-PPase regulation pathways could be used for designing specific inhibitors of this enzyme.
MATERIALS AND METHODS
Genetic constructs. The Mt-PPase/pUC-19 genetic construct for expression of the wild-type Mt-PPase was produced in our previous work [11]. The Arg14Gln mutation was introduced by the two-step PCR with primers 1 (forward) and 2 (reverse) (Evrogen, Russia). Primer sequences are shown in Table 1. The produced fragment was digested with NdeI and BglII restriction endonucleases (Thermo Fisher Scientific, USA) and ligated into linearized pET-42a vector for expression (Thermo Fisher Scientific).
Table 1. Sequences of primers used in the
study
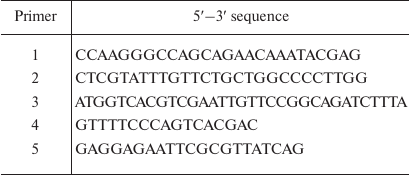
The 1-12 fragment (SLLNVPAGKDLP) from Ec-PPase was attached to the N-terminus of Mt-PPase using previously described plasmids Mt-PPase/pET-28a and NdeI-Ec-PPase/pUC-18 [11]. The latter plasmid contained the PPase gene from E. coli and NdeI recognition site at the 5′-end. Three oligonucleotides were used as primers at different stages (Table 1): primer 3 (encoding amino acid residues 9-13 from Ec-PPase and 1-5 residues from Mt-PPase); primer 4 (M13/pUC reverse primer); and primer 5 (for insertion of EcoRI site at the 3ʹ-end of the Mt-PPase gene). The first polymerase chain reaction was carried out with primers 3 and 4 on the NdeI-Ec-PPase/pUC-18 plasmid (produced in our previous work). The generated fragment was used in the second PCR with the primers 4 and 5 using Mt-PPase/pET-28a plasmid (obtained in our previous work). The produced DNA fragment was hydrolyzed by NdeI and EcoRI restriction endonucleases and ligated into pET23a plasmid.
Purification of proteins. The obtained genetic constructs were expressed in E. coli BL21(DE3) cells, and the recombinant proteins were purified by hydrophobic chromatography as described previously for the wild-type Mt-PPase [11]. Protein yield was 20-30 mg/liter culture. The purity of the enzymes (99%) was evaluated by electrophoresis according to Laemmli [12]. Proteins were identified by mass spectrometry using a MALDI TOF/TOF mass spectrometer (Bruker Daltonics, USA).
Protein preparations were stored at 4°C as suspensions in ammonium sulfate solution (90% saturation). Prior to the use, the enzymes were dissolved in a 50 mM Tris-HCl buffer (pH 7.5) containing 5 mM MgCl2; next, ammonium sulfate was removed by gel filtration on a Sephadex G-50 column (GE Healthcare, USA). Protein concentration was determined spectrophotometrically using the following A2800.1% values calculated from the enzyme amino acid composition using the ProtParam program: 1.04 for the wild-type Mt-PPase and R14Q-Mt-PPase; 0.98 for Ec-Mt-PPase. The theoretical molecular masses of the subunits calculated with the ProtParam program were 18.3 kDa for the wild-type Mt-PPase and R14Q-Mt-PPase and 19.5 kDa for Ec-Mt-PPase.
Pyrophosphate hydrolysis assay. The catalytic activity of PPase was determined from the rate of product (phosphate) formation according to the previously described technique based on the reaction of phosphomolybdate complex with Malachite Green and sodium citrate [13]. An aliquot (0.5-1.5 μl) of PPase solution (total protein concentration, 0.2-0.4 μg/ml) was added to 25 μl of 50 mM Tris-HCl buffer (pH 7.5) containing (if not indicated otherwise) 5 mM Mg2+ and 50 μM MgPPi complex (total PPi concentration, 74.5 μM), and the mixture was incubated at 20°C. At fixed time intervals (15-60 s), the reaction was stopped by adding coloring solution containing Malachite Green (Sigma-Aldrich, USA), ammonium molybdate (Reakhim, Russia), hydrochloric acid (SigmaTek, Russia), polyvinyl alcohol (LenReaktiv, Russia), and deionized water, followed by addition after 15 s of sodium citrate aqueous solution (Sigma-Aldrich). Next, the optical density at 590 nm was measured with a Victor x5 plate reader (Perkin Elmer, USA).
Dependence of Mt-PPase stability on pH and temperature. Mt-PPase solution (0.1 mg/ml) in a buffer (pH 2.6-11) containing 5 mM Mg2+ was incubated for 24 h at 4°C, and the residual activity was measured as described above. The following buffers were used: 50 mM citric acid/50 mM sodium citrate (pH 2.6-4.6); 50 mM MES (pH 5.6-6.6); 50 mM Tris-HCl (pH 7.5-8.6); 50 mM CAPSO (pH 9.6), and 50 mM CAPS (pH 10.6-11). To evaluate the thermal stability of PPase, the enzyme solution (0.1 mg/ml) was incubated for 10 min at 25-75°C in 50 mM Tris-HCl, pH 7.5 (measured at 25°C), containing 5 mM Mg2+, and the residual activity was measured.
Analytical ultracentrifugation was performed in an analytical Spinco E centrifuge (Beckman, USA) at 48,000 rpm with scanning at 280 nm. Prior to centrifugation, enzyme samples were preincubated overnight at 4°C in 50 mM Tris-HCl (pH 7.5) containing 5 mM MgCl2.
Data processing and structure modeling. Inhibition by fluoride and calcium ions, as well as the effect of methylene bisphosphonate (PCP), were described by a simple hyperbolic function. The following scheme was used to describe the bell-shape dependence of the Mt-PPase activity on the concentration of modulating compound.
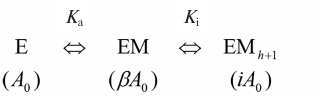
Activation and inhibition of PPase upon sequential binding of modulator ions. M is a modulator (Mg ion, PCP, or allosteric effector); Ka and Ki are apparent dissociation constants of the enzyme complex with the modulator activating and inhibiting ion, respectively; A0 is the activity in the absence of modulator; β and i are activation and inhibition coefficients, respectively. In case of Mg2+ as a modulator, β = ∞, i = 0, because the activity is equal to zero both in the absence of Mg2+ or at the infinitely high Mg2+concentration. Parameters in parentheses designate the activities of the corresponding enzyme forms. The maximal activity corresponds to the EM complexThe reaction rate in the scheme is determined by Eq. (1), where A is the measured activity, h is the empirical coefficient (Hill coefficient analogue). The value of h is less or equal to the number of modulator ions, whose binding causes inhibition, and can assume non-integer values.
A = A0(1 + β[M]/Ka + i[M]h+1/KaKih)/(1 + [M]/Ka + [M]h+1/KaKih). (1)
Equation (2) was used to describe the bell-shaped dependence of PPase stability on pH, which assumed the presence of m and n of basic and acidic groups with pKa1 and pKa2, respectively:
A = Amax/[1 + 10m(pKa1–pH) + 10n(pH–pKa2)]. (2)
The temperature of PPase half-inactivation (T1/2) was determined from the dependence of enzyme activity on the preincubation temperature using Eq. (3), where a is an empiric coefficient:
A = Amax/[1 + (T/T1/2)a]. (3)
SigmaPlot software (Systat Software Inc, USA) was used for the non-linear regression data approximation. Sedimentation coefficient (s20,w) was calculated from the sedimentation profile using the SedFit program [14]. Homology modeling was carried out with the SWISS-MODEL using standard parameters and Ec-PPase structure (PDB ID 1OBW) as a template.
RESULTS
Selection of mutagenesis strategies. The absence of the N-terminal 12-amino acid fragment in Mt-PPase distinguishes it from Ec-PPase and majority of family I PPases [7]. In Ec-PPase, this fragment (SLLNVPAGKDLP) includes several residues that form hydrogen bonds and hydrophobic contacts (Ser1-Ser36, Leu2-Ala38) between the subunits in the enzyme trimer [11]. Due to the lack of this fragment in Mt-PPase, there is a cavity in the intra-trimeric interface, where low-molecular-weight ligands (potential allosteric effectors) can bind.
In order to test this hypothesis, we constructed chimeric Ec-Mt-PPase protein, in which the Mt-PPase polypeptide chain was extended at the N-terminus with residues 1-12 from Ec-PPase. Homology modeling demonstrated that the added fragment could occupy the same position as in Ec-PPase (Fig. 1b) and interfere with the binding of allosteric effector at the intra-trimeric interface.
The Arg14 residue is exposed into the inter-trimeric interface of Mt-PPase (Fig. 1a) and acts as a component of the positively charged Lys11-Arg14-Arg101 cluster formed by residues from three subunits. The network of Arg14 contacts of different nature holds in position the Tyr31–Met36 loop participating in the formation of the inter-trimeric contact. Similar cluster of residues in Ec-PPase is composed of Lys112, Lys115, and Lys146 and located in a different part of the subunit, where it forms an allosteric site that binds ATP (activator) and fructose-1-phosphate (inhibitor) [15, 16].
Purification and characterization of Mt-PPase mutant forms. Two mutant forms of Mt-PPase – R14Q-Mt-PPase and Ec-Mt-PPase – were produced in E. coli. Protein identity was confirmed by MALDI mass spectrometry from the masses of full-length proteins and a set of peptides formed by protein trypsinolysis (data not shown).
The quaternary structure of the three forms of Mt-PPase was evaluated by ultracentrifugation (Fig. 2). In all cases, the major protein fraction was represented by hexamers (Table 2). All protein preparations also contained a small amount of a higher molecular mass form (presumably, dodecamer).
Fig. 2. Sedimentation coefficients for the three forms of Mt-PPase at 20°C. Protein concentration in all experiments was 0.6 mg/ml.
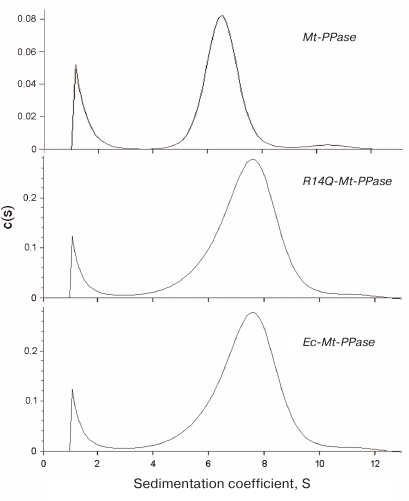
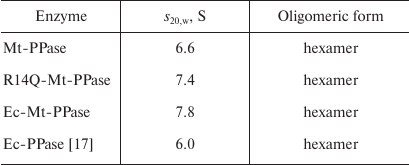
Table 2. Sedimentation coefficients
(s20,w) and suggested oligomeric organization of
PPases
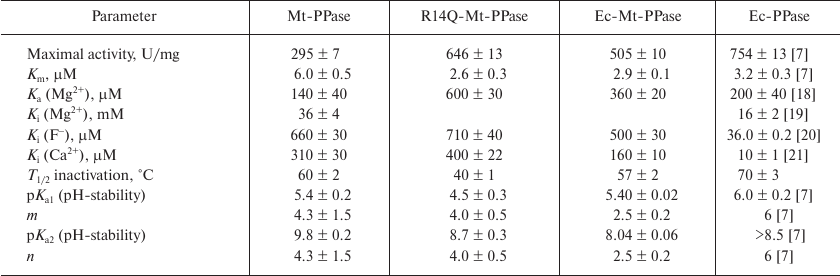
The mutant forms of Mt-PPase were investigated for the catalytic parameters of MgPPi hydrolysis, maximal activity and Km, dissociation constants of complexes with the metal cofactor Mg2+ and inhibitors (Ca2+ and F–), and parameters of pH- and thermal stability of the hexamer (Table 3). The Ka(Mg2+) parameter in Table 3 refers to one of the two Mg2+ ions that activate the enzyme. The second activating Mg2+ ion is bound much stronger and, hence, does not manifest itself over the used range of Mg2+ concentrations. As follows from the obtained data, mutations significantly improved the catalytic characteristics of Mt-PPase. The maximal activity (catalytic constant) increased, while Km decreased, which made these parameters similar to those reported for Ec-PPase. Moreover, the mutations abolished the inhibitory effect of the high concentrations of metal cofactor (Mg2+). On the other hand, both mutants exhibited decreased affinity to the activating Mg2+ ion, and the R14Q mutation reduced the thermal stability of the enzyme. The effects of inhibitors (fluoride and calcium ions) were only slightly altered, except that the chimeric protein demonstrated increased sensitivity to Ca2+ ions.
Table 3. Catalytic properties of Mt-PPase
variants in comparison with Ec-PPase
Activation and inhibition of Mt-PPase with the substrate analogue. The presence of binding sites for the allosteric effector at the subunit contact zones, separate from the active site, is common feature for family I PPases [3]. Some pyrophosphate analogues can bind either in the active site or regulatory allosteric site. In particular, the allosteric regulator ATP can be hydrolyzed in the active site of Ec-PPase [15]. The free form of pyrophosphate activates Ec-PPase via binding at the allosteric site [19, 22]. In order to test the presence of allosteric binding sites in Mt-PPase and their connection to the zones of oligomeric contacts, we investigated the effect of non-hydrolyzable pyrophosphate analogue, PCP, on the activity of Mt-PPase and its mutant variants Ec-Mt-PPase and R14Q-Mt-PPase carrying modifications at the subunit interfaces.
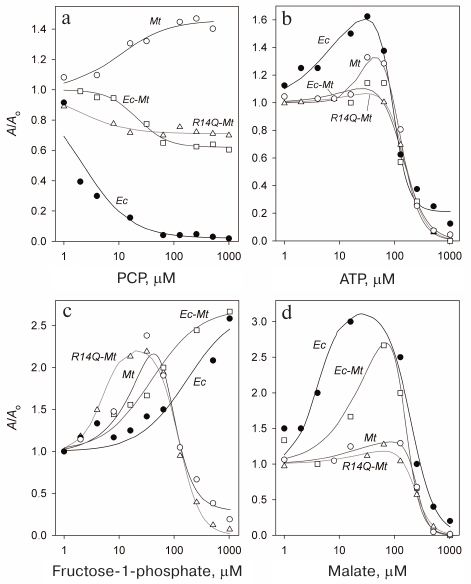
The obtained data (Fig. 3a) demonstrate pronounced distinction in the behavior of Mt-PPase and Ec-PPase. While PCP almost completely inhibited Ec-PPase, Mt-PPase was activated by this compound. Unlike the native enzyme, the mutant forms of Mt-PPase were inhibited by PCP, but the decrease in their activity was no more than 40%.
Fig. 3. Effect of PCP (a), ATP (b), fructose-1-phosphate (c), and L-malate (d) on the activities of Ec-PPase and three forms of Mt-PPase. Mt, Mt-PPase; R14Q-Mt, Mt-PPase with the R14Q substitution; Ec-Mt, Mt-PPase fused with the Ec-PPase fragment; Ec, Ec-PPase. Solid line curves represent the best approximations with Eq. (1).
The obtained dependencies could be described by a simplified scheme suggesting the binding of either activating or inhibiting PCP molecule. The parameters produced using this scheme (Table 4) show that the affinity of the wild-type Mt-PPase to PCP is an order of magnitude lower than that of Ec-PPase. The affinity to PCP virtually did not change for the chimeric Ec-Mt-PPase, but increased almost ten-fold for R14Q-Mt-PPase.
Table 4. Parameters of the interaction of
Ec-PPase and three forms of Mt-PPase with PCP, ATP,
fructose-1-phosphate, and L-malate obtained from the data shown in Fig.
3
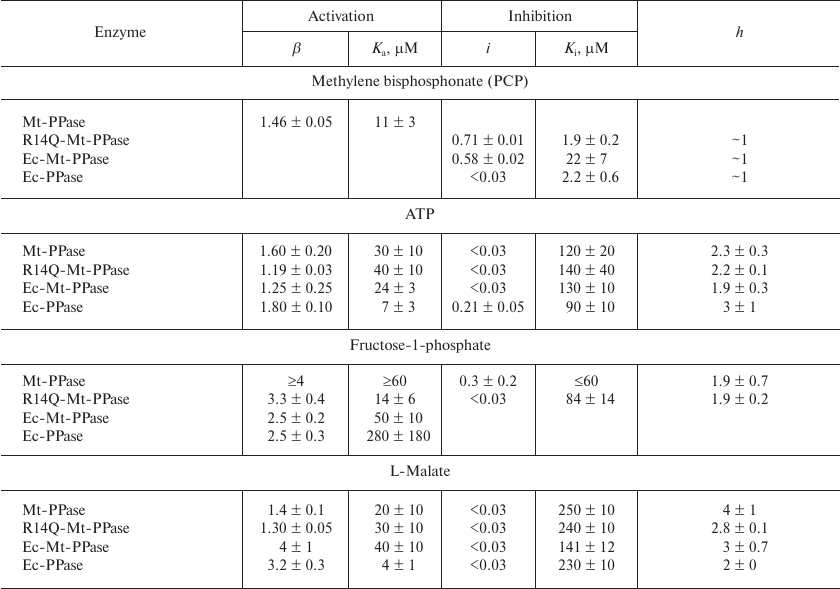
Activation and inhibition of Mt-PPase by metabolites. We also examined the effects of ATP, fructose-1-phosphate, and L-malate on the activity of Mt-PPase. These compounds were selected based on the fact that ATP and fructose-1-phosphate affect the activity of Ec-PPase [15, 16], and bound L-malate was found in the crystal structure of PPase from Bartonella henselae (PDB ID 3SW5). All three compounds activated Mt-PPase at low concentrations and inhibited it at high concentrations (Fig. 3, b-d). The effects of ATP and L-malate was qualitatively similar for all investigated proteins, while the inhibitory effect of fructose-1-phosphate was absent for Ec-PPase and Ec-Mt-PPase. Based on the activity profiles in the presence of fructose-1-phosphate and L-malate, chimeric Ec- Mt-PPase was found to be closer to Ec-PPase than Mt-PPase, while R14Q-Mt-PPase was virtually indistinguishable from the wild-type Mt-PPase (Fig. 3c). The parameters of enzyme activation and inhibition calculated based on the suggested scheme are presented in Table 4. As one can see, the Ka values for different forms of Mt-PPase and various modulators do not differ significantly in the majority of cases. A sharp drop in the activity at high concentrations of ATP and L-malate indicates that this effect is due to the cooperative binding of several inhibitor molecules. The h value equals approximately 2 for ATP and 3 for L-malate. In the majority of other cases, is does not differ significantly from 1.
DISCUSSION
Mutations examined in this work in one way or another are involved into formation of oligomeric contacts in the hexameric Mt-PPase, which explains most of the observed effects. An increased activity of the mutant forms could indicate that formation of oligomer contacts in the native hexameric Mt-PPase suppresses its activity, i.e., represents a form of internal inhibition (“self-inhibition”). Hence, disruption of oligomeric contacts due to the introduced structural changes alleviates this internal inhibition and, in a certain sense, restores catalytic activity of the protein by increasing the catalytic constant and decreasing Km. Furthermore, modification of the subunit contact zones does not change the oligomeric structure of Mt-PPase, but makes it less compact, as shown by the results of sedimentation analysis. In particular, the s20,w values for the mutant forms in Table 2 are significantly higher than for the wild-type enzyme, even when 8% increase in the Ec-Mt-PPase mass is taken into account. In addition to their influence on the catalytic activity, the mutations decreased the enzyme affinity to, and abolished inhibition by, the excess of the metal cofactor ions. Hence, the effect of the cofactor, which also binds to the active site, could depend on the subunit contacts and compactness of the molecule as a whole.
Activation of Mt-PPase by the non-hydrolyzable substrate analogue PCP could indicate that PCP binds to the enzyme region other than the active site. Positive heterotropic cooperativity of the active sites seems like a less probable explanation, because the substrate interacts with the active site in a non-cooperative manner [7, 11]. Our data suggest that the binding site for the activating PCP molecule is either located at the inter-subunit interfaces or interacts through them with the active site, because both mutations removed Mt-PPase activation by PCP. Partial inhibition observed for the mutant forms is also difficult to explain considering that PCP binds to the active site. It is more likely that inhibition develops as the allosteric binding sites get occupied.
In general, the effect of PCP on Mt-PPase is fundamentally different from its effect on Ec-PPase, for which it plays the role of competitive inhibitor with Ki approximately the same as Km [22]. This fact has an important practical implication by suggesting that rational design of selective Mt-PPase inhibitors based on the substrate analogues would not be a very promising approach.
ATP, fructose-1-phosphate, and L-malate are metabolites of key importance for all organisms; hence, their effect on Mt-PPase activity observed in vitro could reflect their participation in the regulation of PPases in vivo. The dissociation constants of enzyme complexes with these effectors are in the range of 10-300 μM, which allows their application as lead compounds in the development of high affinity inhibitors. The effects of ATP and L-malate on Mt-PPase and Ec-PPase are qualitatively similar: both compounds activate these enzymes at low concentrations, but inhibit them at high concentrations (Fig. 3, b and d). This indicates the presence of several binding sites for these metabolites in the protein molecule. Fructose-1-phosphate also exhibited similar dual effect on Mt-PPase, while, in the case of Ec-PPase, it acted only as an activator. The inhibitory effect of fructose-1-phosphate on Mt-PPase was abolished when the N-terminal fragment of Ec-PPase was attached to Mt-PPase. This modification of Mt-PPase also made it similar to Ec-PPase in regard to the effect of PCP.
Taken together, the obtained data indicate the presence of more than one (probably two or three) allosteric effector-binding sites in Mt-PPase. Activation by ligands, as well as partial inhibition by PCP, could not be explained by the binding of these compounds in the active site. The respective allosteric sites (conditionally named “activating sites”) could be located at the inter-trimeric interfaces. This follows from the data of X-ray diffraction analysis of other hexameric PPases, in which bound L-malate and phosphate were found only in this zone (PDB ID 3SW5 and 3FQ3, respectively), which implies that the respective site exhibits the highest affinity to these ligands. The affinity of the “inhibitory site” to ATP, fructose-1-phosphate, and L-malate is significantly lower, but their binding at these sites is characterized by high cooperativity. The observed regularities can be explained within the framework of the model according to which both activating and inhibiting ligands bind to the same allosteric site located at the inter-subunit interface of each subunit. The filling of this center in one subunit causes enzyme activation, while its filling in other subunits causes inhibition, which implies interconnection between the subunits.
An alternative explanation suggests the binding of effectors at two (or even three) allosteric sites in each subunit. One of these sites is activating, and the others are inhibitory. If the subunit has one inhibitory site, the cooperative nature of inhibition can be explained only by interaction between the inhibitory sites in different subunits. Considering that the cavity at the inter-trimeric interface is likely the activating site, the role of inhibitory sites could be assigned to the cavity that is exposed in Mt-PPase due to the lack of the N-terminal fragment. The absence of inhibition of Ec-Mt-PPase by fructose-1-phosphate (similarly to the Ec-PPase) supports this suggestion. If two inhibitory sites are present, they can be independent. Moreover, one of the inhibitory sites could act as the enzyme active site. The possibility of ATP binding in the active site follows from the presence of ATPase activity in Mt-PPase [11]. Our further studies are aimed at experimental verification of the above possibilities.
Acknowledgements. The authors are grateful to P. V. Kalmykov and N. N. Magretova for performing analytical ultracentrifugation, N. N. Vorobyeva for providing Ec-PPase, and M. V. Serebryakov for performing protein mass spectrometry analysis. MALDI-mass spectrometer was used in the framework of the Program of Moscow State University Development.
Conflict of interest. The authors declare no conflict of interest in financial or any other sphere.
Ethical approval. This article does not contain description of studies with human participants or animals performed by any of the authors.
REFERENCES
1.Baykov, A. A., Cooperman, B. S., Goldman, A., and
Lahti, R. (1999) Cytoplasmic inorganic pyrophosphatase, in Inorganic
Polyphosphates, Springer, pp. 127-150.
2.Cooperman, B. S., Baykov, A. A., and Lahti, R.
(1992) Evolutionary conservation of the active site of soluble
inorganic pyrophosphatase, Trends Biochem. Sci., 17,
262-266.
3.Samygina, V. R. (2016) Inorganic pyrophosphatases:
structural diversity serving the function, Russ. Chem. Rev.,
85, 464-476.
4.Heikinheimo, P., Tuominen, V., Ahonen, A.-K.,
Teplyakov, A., Cooperman, B., Baykov, A., Lahti, R., and Goldman, A.
(2001) Toward a quantum-mechanical description of metal-assisted
phosphoryl transfer in pyrophosphatase, Proc. Natl. Acad. Sci.
USA, 98, 3121-3126.
5.Vijayan, M. (2005) Structural biology of
mycobacterial proteins: the Bangalore effort, Tuberculosis
(Edinb), 85, 357-366.
6.Rodina, E. V., Valueva, A. V., Yakovlev, R. Y.,
Vorobyeva, N. N., Kulakova, I. I., Lisichkin, G. V., and Leonidov, N.
B. (2015) Immobilization of inorganic pyrophosphatase on nanodiamond
particles retaining its high enzymatic activity, Biointerphases,
10, 041005.
7.Tammenkoski, M., Benini, S., Magretova, N. N.,
Baykov, A. A., and Lahti, R. (2005) An unusual, His-dependent family I
pyrophosphatase from Mycobacterium tuberculosis, J. Biol.
Chem., 280, 41819-41826.
8.Pratt, A. C., Dewage, S. W., Pang, A. H., Biswas,
T., Barnard-Britson, S., Cisneros, G. A., and Tsodikov, O. V. (2015)
Structural and computational dissection of the catalytic mechanism of
the inorganic pyrophosphatase from Mycobacterium tuberculosis,
J. Struct. Biol., 192, 76-87.
9.Harutyunyan, E. H., Oganessyan, V. Y., Oganessyan,
N. N., Avaeva, S. M., Nazarova, T. I., Vorobyeva, N. N., Kurilova, S.
A., Huber, R., and Mather, T. (1997) Crystal structure of holo
inorganic pyrophosphatase from Escherichia coli at 1.9 Å
resolution. Mechanism of hydrolysis, Biochemistry, 36,
7754-7760.
10.Pettersen, E. F., Goddard, T. D., Huang, C. C.,
Couch, G. S., Greenblatt, D. M., Meng, E. C., and Ferrin, T. E. (2004)
UCSF Chimera – a visualization system for exploratory research
and analysis, J. Comput. Chem., 25, 1605-1612.
11.Rodina, E. V., Vainonen, L. P., Vorobyeva, N. N.,
Kurilova, S. A., Sitnik, T. S., and Nazarova, T. I. (2008) Metal
cofactors play a dual role in Mycobacterium tuberculosis
inorganic pyrophosphatase, Biochemistry (Moscow), 73,
897-905.
12.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685.
13.Rowlands, M. G., Newbatt, Y. M., Prodromou, C.,
Pearl, L. H., Workman, P., and Aherne, W. (2004) High-throughput
screening assay for inhibitors of heat-shock protein 90 ATPase
activity, Anal. Biochem., 327, 176-183.
14.Schuck, P., Gillis, R. B., Besong, T. M.,
Almutairi, F., Adams, G. G., Rowe, A. J., and Harding, S. E. (2014)
SEDFIT–MSTAR: molecular weight and molecular weight distribution
analysis of polymers by sedimentation equilibrium in the
ultracentrifuge, Analyst, 139, 79-92.
15.Rodina, E. V., Vorobyeva, N. N., Kurilova, S. A.,
Sitnik, T. S., and Nazarova, T. I. (2007) ATP as effector of inorganic
pyrophosphatase of Escherichia coli. The role of residue Lys112
in binding effectors, Biochemistry (Moscow), 72,
100-108.
16.Vorobyeva, N. N., Kurilova, S. A., Anashkin, V.
A., and Rodina, E. V. (2017) Inhibition of Escherichia coli
inorganic pyrophosphatase by fructose-1-phosphate, Biochemistry
(Moscow), 82, 953-956.
17.Vainonen, Yu. P., Kurilova, S. A., and Avaeva, S.
M. (2002) Hexameric, trimeric, dimeric, and monomeric forms of
inorganic pyrophosphatase from Escherichia coli, Russ. J.
Bioorg. Chem , 28, 385-391.
18.Avaeva, S. M., Rodina, E. V., Kurilova, S. A.,
Nazarova, T. I., Vorobyeva, N. N., Harutyunyan, E. H., and Oganessyan,
V. Y. (1995) Mg2+ activation of Escherichia coli
inorganic pyrophosphatase, FEBS Lett., 377, 44-46.
19.Sitnik, T., Vainonen, J., Rodina, E., Nazarova,
T., Kurilova, S., Vorobyeva, N., and Avaeva, S. (2003) Effectory site
in Escherichia coli inorganic pyrophosphatase is revealed upon
mutation at the intertrimeric interface, IUBMB Life, 55,
37-41.
20.Samygina, V. R., Moiseev, V. M., Rodina, E. V.,
Vorobyeva, N. N., Popov, A. N., Kurilova, S. A., Nazarova, T. I.,
Avaeva, S. M., and Bartunik, H. D. (2007) Reversible inhibition of
Escherichia coli inorganic pyrophosphatase by fluoride: trapped
catalytic intermediates in cryo-crystallographic studies, J. Mol.
Biol., 366, 1305-1317.
21.Avaeva, S. M., Vorobyeva, N. N., Kurilova, S. A.,
Nazarova, T. I., Polyakov, K. M., Rodina, E. V., and Samygina, V. R.
(2000) Mechanism of Ca2+-induced inhibition of
Escherichia coli inorganic pyrophosphatase, Biochemistry
(Moscow), 65, 373-387.
22.Vainonen, J. P., Vorobyeva, N. N., Rodina, E. V.,
Nazarova, T. I., Kurilova, S. A., Skoblov, J. S., and Avaeva, S. M.
(2005) Metal-free PPi activates hydrolysis of
MgPPi by an Escherichia coli inorganic
pyrophosphatase, Biochemistry (Moscow), 70, 69-78.