An Assay for the Activity of Base Excision Repair Enzymes in Cellular Extracts Using Fluorescent DNA Probes
O. A. Kladova1, D. A. Iakovlev1, R. Groisman2,3, A. A. Ishchenko2,3, M. K. Saparbaev2,3, O. S. Fedorova1,4,a*, and N. A. Kuznetsov1,4,b*
1Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia2Groupe “Réparation de l'AND”, Equipe Labellisée par la Ligue Nationale contre le Cancer, CNRS UMR 8200, Université Paris-Sud, Université Paris-Saclay, F-94805 Villejuif, France
3Gustave Roussy, Université Paris-Saclay, F-94805 Villejuif, France
4Novosibirsk State University, 630090 Novosibirsk, Russia
* To whom correspondence should be addressed.
Received January 21, 2020; Revised February 13, 2020; Accepted February 17, 2020
Damaged DNA bases are removed by the base excision repair (BER) mechanism. This enzymatic process begins with the action of one of DNA glycosylases, which recognize damaged DNA bases and remove them by hydrolyzing N-glycosidic bonds with the formation of apurinic/apyrimidinic (AP) sites. Apurinic/apyrimidinic endonuclease 1 (APE1) hydrolyzes the phosphodiester bond on the 5′-side of the AP site with generation of the single-strand DNA break. A decrease in the functional activity of BER enzymes is associated with the increased risk of cardiovascular, neurodegenerative, and oncological diseases. In this work, we developed a fluorescence method for measuring the activity of key human DNA glycosylases and AP endonuclease in cell extracts. The efficacy of fluorescent DNA probes was tested using purified enzymes; the most efficient probes were tested in the enzymatic activity assays in the extracts of A549, MCF7, HeLa, WT-7, HEK293T, and HKC8 cells. The activity of enzymes responsible for the repair of AP sites and removal of uracil and 5,6-dihydrouracil residues was higher in cancer cell lines as compared to the normal HKC8 human kidney cell line.
KEY WORDS: enzymatic activity, fluorescence, DNA probe, DNA glycosylase, AP-endonucleaseDOI: 10.1134/S0006297920040082
Abbreviations: AAG, alkyladenine DNA glycosylase; APE1, human AP endonuclease 1; AP site, apurinic/apyrimidinic site; BHQ, black hole quencher; DHU, 5,6-dihydrouridine; εA, 1,N6-ethenoadenosine; FAM, 6-carboxyfluorescein; FRET, Förster resonance energy transfer; F, (2R,3S)-2-(hydroxymethyl)-3-hydroxytetrahydrofuran residue; MBD4, methyl-CpG-binding domain 4; NEIL1, human endonuclease VIII; NTHL1, human endonuclease III; OGG1, 8-oxoguanine DNA glycosylase; oxoG, 8-oxoguanosine; TDG, thymine DNA glycosylase; UDG, uracil-DNA glycosylase.
Oxidation, alkylation, deamination, formation of apurinic/apyrimidinic
(AP) sites, and DNA strand breaks are on the list (although incomplete)
of processes that lead to the damage of DNA structure [1-7]. On one hand, DNA damage can
initiate malignant cell transformation; on the other hand, the same
types of DNA damage occur during cancer chemotherapy and radiotherapy
[8-10]. Enzymatic DNA repair
system plays an important role in the maintenance of normal functioning
of living organisms. It has been shown in numerous studies that the
activity of DNA repair enzymes affects cell susceptibility to
genotoxins and/or damaging factors, including those used in cancer
chemotherapy and radiotherapy [11, 12]. Functional disruption of DNA repair enzymes has
severe consequences in the human body and often leads to cancer
development and premature aging [13, 14]. Base excision repair (BER) is one of the
enzymatic mechanisms used by the cells to remove and replace modified
and incorrectly paired bases in DNA. BER is initiated by DNA
glycosylases, which recognize modified bases and catalyze hydrolysis of
the N-glycosidic bond [15, 16]. Next, apurinic/apyrimidinic (AP) endonuclease
removes the remaining fragment of 2′-deoxyribose. The major role
of these enzymes is quick and accurate recognition of modified base or
AP site among numerous unmodified bases and initiation of DNA repair
process.
Cells and animals deficient by DNA glycosylases are more sensitive to the effects of DNA-damaging factors [17-19]. At the same time, abolishing AP endonucleases from the cells by genetic engineering methods leads to cell death, indicating the critical role of these enzymes in the restoration of intact DNA structure [20-22].
The methods for determining the activity of DNA repair enzymes in cell extracts have been intensively developed [23-29]. Several different approaches have been proposed for assaying the activity of DNA glycosylases that usually involve the recording of fluorescent signal generated by DNA probes containing a damaged nucleotide specific for the studied enzyme [30-32]. Typically, a pair of dyes capable of Förster resonance energy transfer (FRET) is used as signal source. Other assays are based on the difference in the fluorescence intensity of fluorescent analogues of bases incorporated into single- or double-stranded DNA probes [33]. For example, Gines et al. [23] modified DNA probes with biotin for immobilization on magnetic nanoparticles. The reaction mixture containing DNA probe and different amounts of alkyladenine DNA glycosylase (AAG) and/or human AP endonuclease 1 (APE1) was incubated for 1 h, after which its fluorescence intensity was recorded. The authors found that the nanoparticles degraded at a rate of 2% per day [23]; hence, the use of these method was complicated by the necessity for verification of the nanoparticle quality and control of the fluorescent DNA probe concentration on the nanoparticle surface. Hu et al. [30] used special equipment to analyze the activities of 8-oxoguanine DNA glycosylase (OGG1) and AAG at a single-molecule level. The reaction mixture was incubated for 1.5 h, and then the specific activity of the enzyme was determined from the fluorescent signal produced by the products formed in the reaction. Recently, a new sensitive method for detecting uracil-DNA glycosylase (UDG) activity based on the terminal deoxynucleotidyl transferase-assisted formation of fluorescent copper nanoclusters was reported [34]. Several other approaches for signal amplification in the UDG activity assay were also described [35-37]. Despite the abundance of options for determination of enzymatic activity in cell extracts, some methods mentioned above involve a large number of steps and require special equipment and use of other enzymes, such as DNA polymerase, DNA nickase, ribonuclease, and even AP endonuclease.
It should be also emphasized that most activity assays are based on comparing intensities of the fluorescent signal in the reaction mixture after a certain period of time and the signal obtained using purified enzyme. However, numerous studies [38-43] have demonstrated that the activity of DNA repair enzymes changes significantly when these enzymes interact with other proteins and enzymes that can be present in the cell extract but not in standard samples used for activity quantification. Therefore, the same amount of the enzyme will cause different changes in the fluorescent signal in the control sample and cell extract.
In this study, we tested a FRET-based assay for the activity of key BER enzymes (human DNA glycosylases and AP endonuclease) in the cell extracts of different human cell lines. The main goal of this work was to develop an easy method for measuring the activity of BER enzymes in cell extracts. The advantage of the developed assay over other approaches is its simplicity. The method uses a small number of cells and synthetic FRET-labeled DNA duplex containing damaged nucleotide in its middle part. The novelty of our method is that it allows to analyze both the signal amplitude after a certain period of time and the rate of DNA probe cleavage.
Synthesized fluorescent DNA probes contained modified nucleotides, such as (2R,3S)-2-(hydroxymethyl)-3-hydroxytetrahydrofuran residues (F), 5,6-dihydrouridine (DHU), 1,N6-ethenoadenosine (εA), 8-oxoguanosine (oxoG), or uridine (Table 1). The efficacy of each probe was tested with purified human DNA glycosylases and AP endonuclease. The most efficient probes were used to determine the activity of BER enzymes in the extracts of A549, MCF7, HeLa, WT-7, HEK293T, and HKC8 cells. The same probes containing no damaged nucleotides were used a control for nonspecific cleavage to estimate the sensitivity of the probes to nonspecific endo- and exonucleases in the cell extracts. We showed that the tested fluorescent DNA probes can be used for measuring the activity of several human DNA glycosylases and AP endonuclease. The assay sensitivity was sufficient to measure the activity of enzymes in the extracts from 106 cells with a high reproducibility. Analysis of different cell lines revealed the differences in the activity of key BER enzymes in tumor and non-tumor cells.
Table 1. DNA probes used as substrates for
DNA glycosylases and AP endonuclease

Notes: FAM, 6-carboxyfluorescein; BHQ1, black hole quencher
(fluorescence quencher).
MATERIALS AND METHODS
The structure of DNA probes. DNA probes were oligonucleotide duplexes containing the FRET pair of dyes [6-carboxyfluorescein (FAM) and black hole quencher (BHQ1)] and damaged nucleotide specific for the studied enzyme or a group of enzymes (Table 1). The removal of the damaged nucleotide resulted in the formation of either the AP site [in the case of monofunctional DNA glycosylases, such as UNG2, SMUG1, methyl-CpG-binding domain 4 (MBD4), thymine DNA glycosylase (TDG), and AAG] or a single-strand break in the DNA strand containing this nucleotide [in the case of bifunctional DNA glycosylases, such as OGG1, human endonuclease VIII (NEIL1), and human endonuclease III (NTHL1)]. APE1 also generated a single-strand break in the strand containing the F residue. After formation of the gap, short oligonucleotide fragments were released from the DNA duplex product, thus leading to the increase in the distance between FAM and BHQ1 and, therefore, significant increase in the FAM fluorescent signal.
Cell procedures. The activity of DNA repair enzymes was assayed in the A549, HeLa, MCF7, WT-7, HEK293T, and HKC8 cells (Table 2). HEK293T, A549, WT-7, HeLa, and HKC8 cells were cultured in DMEM (Gibco, Thermo Fisher Scientific, USA) and MCF7 cells were cultured in RPMI-1640 medium (Gibco, Thermo Fisher Scientific) in a humidified atmosphere containing 5% CO2 at 37°C. Both media were supplemented with 10% fetal calf serum (Gibco, Thermo Fisher Scientific), 100 μg/ml streptomycin, and 100 U/ml penicillin.
Table 2. Human cell lines used in this
work
The cells were mechanically detached from the culture flask surface, counted, pelleted by centrifugation (1000g, 2 min), and washed with PBS twice before lysis. The cells were counted with a Countess II Automated Cell Counter (Thermo Fisher Scientific). For this, 10 μl of the cell suspension was mixed with 10 μl of trypan blue solution, and 10 μl of the resulting mixture was placed in a cell counting cassette (Bio-Rad, USA).
Preparation of cell extracts. To obtain cell extracts for the enzyme activity assay, 106 cells were lysed in 200 μl of CHAPS buffer (10 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 1 mM EDTA, 0.5% CHAPS, 10% glycerol, 0.1 mM PMSF, and 0.5 mM β-mercaptoethanol). Total protein concentration was measured by the Bradford method (typically, 1-2 μg/μl). The cell extracts were stored at —80°C and thawed immediately before the experiment.
Enzyme activity assays in the cell extracts. The reaction mixture was prepared in 100 μl of the reaction buffer (50 mM Tris-HCl, pH 7.5, 50 mM KCl, 1 mM EDTA, 1 mM DTT, and 7% glycerol) and contained 70 μl of the cell extract. DNA probe with specific damaged nucleotide (Table 1) was added to the cell extract to the final concentration of 1 μM. After rapid mixing, FRET signal was recorded using a FLUOstar Omega fluorometer (BMG Labtech, Germany) at the excitation wavelength (λex) of 485 nm and emission wavelength (λem) of 520 nm. The reaction was carried out at 37°C; the maximum reaction duration was 2000 s. Each experiment was repeated three times. The relative activity of the enzymes in different cell lines was compared using the amplitude of the FRET signal after incubation of the reaction mixture for 2000 s.
Kinetic analysis of recombinant enzymes by the stopped-flow fluorescence method. The pre-steady-state kinetics of the recombinant enzymes was studied with an SX.18MV stopped-flow spectrometer (Applied Photophysics Ltd., UK) by monitoring the FRET-generated fluorescence signal as described earlier [44-47]. The DNA probes were modified with FAM and BHQ1 (Table 1); the excitation wavelength (λex) was 494 nm; fluorescence was recorded at λem > 515 nm (Schott filter OG-515). The instrument’s dead time was 1.4 ms. All experiments were conducted at 25°C in the buffer containing 50 mM Tris-HCl, pH 7.5, 50 mM KCl, 1 mM EDTA, 1 mM DTT, 5 mM MgCl2, and 7% glycerol (v/v).
DNA glycosylase or DNA glycosylase/APendonuclease mixture in the buffer were placed in one of the instrument’s syringes and rapidly mixed in the reaction chamber with the DNA probe solution in the buffer injected with another syringe. The concentrations of the enzymes and DNA probe were 1 μM in all the experiments. The reported concentrations of reactants are those in the reaction chamber after mixing. Typically, the kinetic curves shown in the figures were averaged from four or more fluorescence traces recorded in individual experiments. Changes in the FRET signal corresponded to the changes in the distance between the dye and the quencher caused by conformational distortion of the DNA helix during formation of the enzyme–DNA complex and subsequent DNA cleavage.
RESULTS AND DISCUSSION
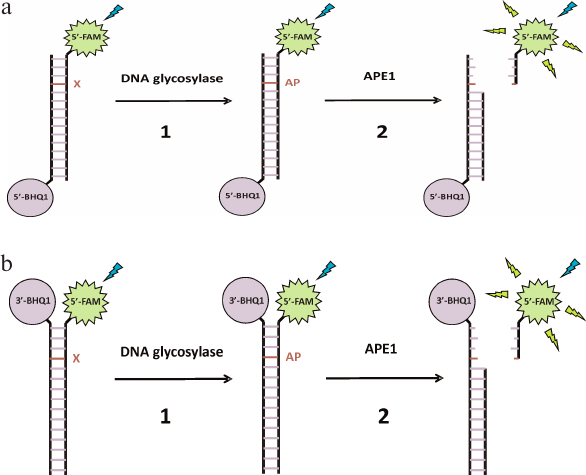
To determine the catalytic activity of DNA glycosylases and AP endonuclease, two types of FRET-labeled DNA substrates were designed that contained damaged nucleotides specific for the studied enzymes (Fig. 1). Type I DNA substrates (Fig. 1a) contained FAM (emitter) and BHQ1 (quencher) at the 5′-ends of the oligonucleotides; type II DNA substrates (Fig. 1b) contained FAM at the 5′-end and BHQ1 at the 3′-end of the duplex-forming oligonucleotides.
Fig. 1. Interaction of monofunctional DNA glycosylases with type I (a) and type II (b) FRET-labeled substrates. Stage 1, removal of damaged base (X) and formation of AP site; stage 2: cleavage of AP site by APE1, leading to the release of short oligonucleotide fragment from the product duplex and increase in the distance between FAM (fluorophore) and BHQ1 (quencher) accompanied by a significant increase in the FRET signal.
It should be noted that analysis of the activity of monofunctional DNA glycosylases that form an AP site in the DNA duplex is more complicated compared to bifunctional DNA glycosylases or AP endonuclease, which form single-strand breaks in the DNA substrates. Therefore, the sensitivity of both types of DNA substrates was analyzed using monofunctional MBD4 and TDG DNA glycosylases (Fig. 2). Interaction of MBD4 and TDG with model uracil-containing DNA substrates led to the uracil removal and formation of the AP site (Fig. 1, stage 1). This process was accompanied by a slow increase in the FRET signal (Fig. 2a). It has been demonstrated earlier for MBD4 that the slow growth in the FRET signal within 100-3000 s corresponds to slow accumulation of reaction products and presumably characterizes the rate-limiting dissociation of the enzyme complex with the reaction product containing the AP site [48].
Fig. 2. Changes in the FRET signal resulting from the interaction of MBD4 and TDG with DNA substrates containing uridine as a damaged nucleotide. a) The sensitivity of type I and type II DNA substrates to MBD4 and TDG. b) Effect of APE1 on the interaction of MBD4 and TDG with the type II substrate.
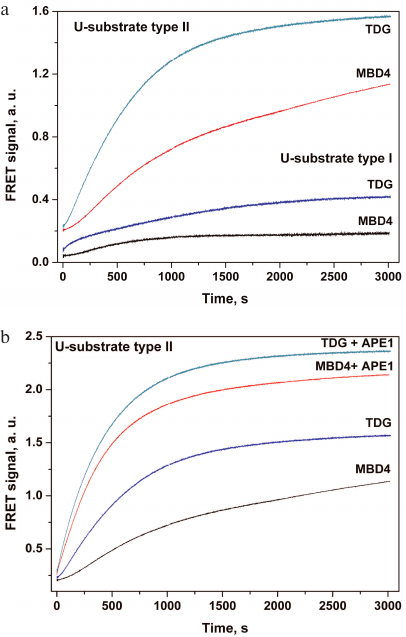
Type II DNA substrate (Fig. 1b) containing FRET labels at the same end of the DNA duplex should provide a greater change in the amplitude of the FRET signal because of greater difference in the distance between the emitter and quencher molecules before and after catalysis, and therefore should demonstrate higher sensitivity to MBD4 and TDG. Therefore, this model substrate was used in subsequent experiments involving APE1. The interaction of MBD4 and TDG with this substrate in the presence of APE1 led to an approximately 2-fold increase in the FRET signal amplitude (Fig. 2b), indicating formation of a single-strand break at the AP site catalyzed by AP endonuclease. It has been shown previously that APE1 stimulates the activity of DNA glycosylases under conditions of multiple enzyme turnover [38-43]. Therefore, the presence of AP endonuclease in the reaction mixture promotes change in the FRET signal amplitude during interaction of the damaged DNA with monofunctional DNA glycosylases and increases the sensitivity of probes to these enzymes.
We found that the sensitivity of type I DNA probes to enzymatic cleavage was low, but still sufficient to detect this process in vitro. However, even such low sensitivity allowed registration of the AP site formation accompanied by the increase in the flexibility of DNA duplex after uracil removal by monofunctional DNA glycosylases and decrease in the distance between the dyes. Type II DNA probes were more sensitive to the cleavage because of the shorter distance between the fluorophore and the quencher. In the case of simultaneous action of DNA glycosylase and AP endonuclease in cell extracts, the change in the FRET signal is additionally doubled (Fig. 2b). Therefore, we used more sensitive type II DNA substrates to evaluate the enzymatic activity in cell extracts containing both enzymes.
To determine the activity of the DNA repair enzymes, we used type II DNA substrates (Table 1) containing enzyme-specific damaged nucleotides. Such nucleotides can appear in the DNA under the action of various environmental factors, e.g., oxidative stress (oxoG and DHU), deamination (uridine), alkylation (εA), and those inducing formation of the AP sites. Using a set of DNA probes containing these lesions, we characterized the activity of all key human DNA repair enzymes involved in the removal of damaged bases from DNA. For OGG1, the probe contained an oxoG (oxoG-probe); for UNG2, SMUG1, MBD4, and TDG (enzyme that specifically recognize uracil in DNA), the probes contained uridine (U-probes); for NEIL1 and NTHL1, the DNA probe contained a DHU residue (DHU-probe); for AAG, the DNA duplexes contained εA (εA-probe). The DNA probe with the AP site analog (2R,3S)-2-(hydroxymethyl)-3-hydroxytetrahydrofuran residue (F-probe) was used as a substrate for APE1. Whole-cell extracts contain multiple exo- and endonucleases that can degrade DNA probes; therefore, to rule out the nonspecific cleavage of DNA probes, we used the DNA duplex that did not contain damaged nucleotides (C-probe) (Table 1).
The activity of DNA repair enzymes was analyzed in human tumor cell lines of different histological origin (Table 2). The reaction mixture contained the whole-cell extract and DNA probe with the damaged nucleotide. Figure 3 shows kinetic curves for the DNA probe cleavage in the extracts of studied cell lines. For all the cell lines, the cleavage of DNA probes containing F, uridine, or DHU residues continued up to 2000 s and was accompanied by a significant increase in the amplitude of FRET signal associated with the activity of the corresponding enzymes (Table 1). It should be noted that the cleavage of the intact duplex (C-probe) was accompanied by a slight increase in the FRET signal, which pointed to much slower nonspecific DNA cleavage. Various approaches are used to protect DNA probes against cleavage by exonucleases, including hairpin structures and modified terminal nucleotides or phosphate groups. However, the use of a small number of cells allowed us to neglect the contribution of nonspecific DNA probe cleavage during the analysis. By comparing the extent of specific and nonspecific DNA cleavage, we were able to conclude that in the case of F-, U-, and DHU-probes, probe degradation by cell nucleases can be neglected and, therefore, standard oligonucleotide probes can be used without introducing additional protective structures into them.
Fig. 3. Changes in the FRET signal characterizing the activity of DNA repair enzymes in the extracts of A549 (a), MCF7 (b), HeLa (c), WT-7 (d), HEK293T (e), and HKC8 (f) cell lines. See Table 1 for the description of the used probes.
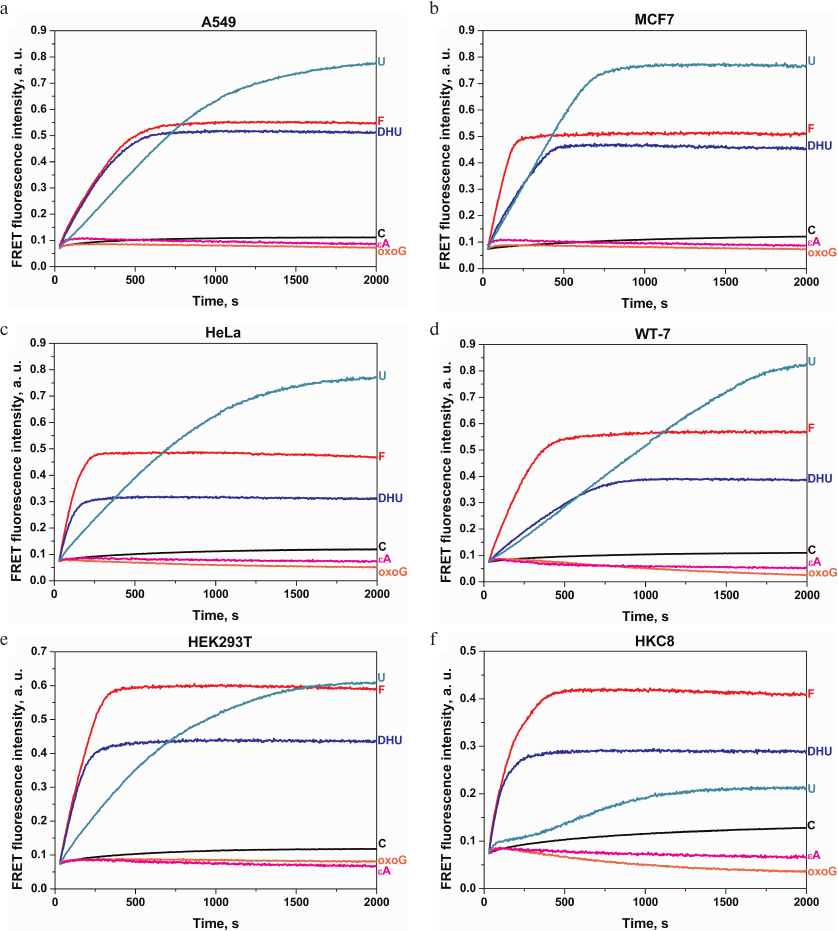
In contrast to the F-, U-, and DHU-probes, processing of oxoG- and εA-probes (cleaved by OGG1 and AAG, respectively) was accompanied by a short-time increase in the FRET signal in the initial part of the kinetic curve followed by the signal amplitude decrease within entire time interval up to 2000 s in all cell lines. Such changes in the FRET signal could not be explained by direct probe cleavage (Fig. 3) or nonspecific DNA degradation, since in both these cases, DNA cleavage should be accompanied by an increase in the FRET signal (Fig. 1b), but rather indicate more complex interactions of these DNA probes with the enzymes in the cell extracts or suggest that the concentration of OGG1 and AAG in the cell extracts obtained from 106 cells was insufficient concentrations for activity detection. Therefore, it is possible that the concentrations of OGG1 and AAG (the only enzymes responsible for the removal of oxoG and alkylated bases, respectively) in the cell extracts were significantly lower than the concentrations of uracil-DNA glycosylases UNG2, SMUG1, MBD4, and TDG and DNA glycosylases NEIL1 and NTHL1 responsible for the removal of damaged pyrimidine nucleotides.
The relative activities of the enzymes in the extracts of different cell lines were compared by monitoring the changes in the FRET signal amplitude 2000 s after the reaction initiation. As shown in Fig. 4a, insignificant nonspecific cleavage of the C-probe took place in all cell lines. A more pronounced increase in the FRET signal was observed for the F-, U-, and DHU-probes. Comparison of the FRET signal amplitudes for the F-probe revealed only small differences between the cell lines (e.g., less than a 1.5-fold difference between the normal human cell lines HKC8 and HEK293T). Therefore, the level of AP endonuclease activity varied only slightly between the studied cell lines.
Fig. 4. Relative activities of DNA repair enzymes in the extracts of A549, MCF7, HeLa, WT-7, HEK293T, and HKC8 cells: a) FRET signal; b) initial slopes of the kinetic curve.
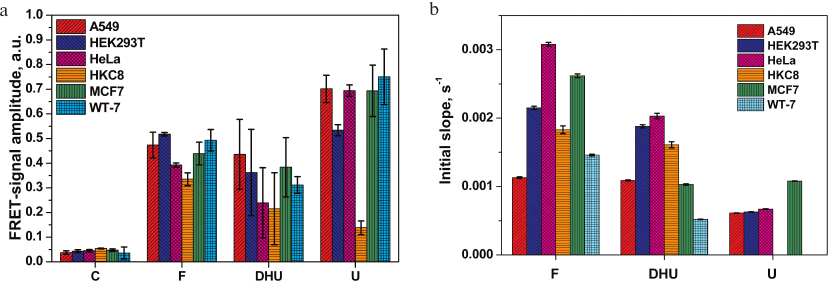
The differences in the amplitude of the FRET signal for the DHU-probe cleavage by NEIL1 and NTHL1 were more significant (e.g., ~2-fold difference was observed for the HKC8 and A549 cells). Analysis of total uracil-DNA glycosylase activity of UNG2, SMUG1, MBD4, and TDG revealed a 5-fold increase in the FRET signal amplitude in all cancer cell lines vs. normal human kidney cells HKC8. In contrast, the level of uracil-DNA glycosylase activity in HEK293T cells was only 3.8-times higher than in HKC8 cells. It should be noted that HKC8 cells demonstrated the lowest DNA repair activity with all the DNA probes used.
The rate of DNA cleavage, which is another relevant variable for the comparing enzymatic activities in different extracts, was estimated as the initial slope of the kinetic curve (Fig. 4b) in the case of steady-state kinetic study of DNA cleavage. However, due to complex protein–protein interactions between the enzymes involved in the initial steps of BER, mutual influence of these enzymes on each other’s activity, and the presence of several enzymes with the same substrate specificity in the cell extract, the increase in the FRET signal generated by the cleavage of U-probes in the HKC8 cell extract could not be described by the steady-state kinetics (Fig. 3f), which prevented us from using the initial slope of the kinetic curve in this case. In all other cases, comparison of initial slopes of kinetic curves revealed that the rate of the F-site cleavage varied no more than 3-fold, with the highest and the lowest values observed for the A549 and HeLa cell extracts, respectively. The maximal (4-fold) difference between the activities toward the DHU-probes was found for extracts of WT-7 and HeLa cells. HKC8 cells displayed an intermediate activity against F and DHU. Because of the non-steady-state type of kinetics of cleavage of the U-probe in the HKC8 cell extract, the apparent rate constant of DNA cleavage was not calculated. Nevertheless, the difference in the activity of uracil-DNA glycosylases between the other cell lines did not exceed the factor of two.
In this study, we developed and verified a method for assaying the activities of human BER enzymes in the extracts of HKC8, WT-7, A549, MCF7, HEK293T, and HeLa cells using fluorescent probes. For this purpose, a set of fluorescent DNA probes was designed that contained various types of damaged nucleotides, including those occurring during chemotherapy and radiation therapy of cancer. Using these probes, we were able to estimate relative activities of human DNA glycosylases and AP endonuclease in normal cells and tumor cell lines. It was shown that the nonspecific degradation in the cell extracts of DNA probes containing damaged nucleotides was much slower than specific DNA cleavage. The activity of DNA repair enzymes processing F-, DHU-, and U-probes was higher in cancer cell lines that in the normal human kidney cell line HKC8.
These results can be used for creating a statistical library of relative activities of DNA repair enzymes in cancer cell lines and human tumor tissue cells. In the future, such molecular characterization of cells can help to elucidate the mechanisms underlying tumorigenesis and cancer progression, as well as cell response to some therapies.
Funding. This work was supported by the Russian Science Foundation (project no. 18-14-00135, design of DNA probes and assays in cell lines; project no. 16-14-10038, measuring relative activities of purified MBD4 and TDG); Russian Ministry of Science and Education (project no. AAAA-A17-117020210022-4 for O.S.F. and N.A.K.); Ligue National Contre le Cancer “Equipe Labellisee”, Electricité de France (RB 2017 to M.S.); French National Research Agency (ANR-18-CE44-0008); and Foundation ARC (PJA-20181208015 to A.A.I.).
Authors’ contribution. O.A.K., D.A.I., and R.G. conducted the experiments; O.A.K., N.A.K., and O.S.F. analyzed the data; A.A.I., M.K.S., N.A.K., and O.S.F. contributed reagents, materials, and/or analytical tools; O.A.K., N.A.K., and O.S.F. wrote the paper.
Conflict of interest. The authors declare no conflict of interest.
Compliance with ethical norms. This article does not contain studies with human participants or animals performed by any of the authors.
REFERENCES
1.Wallace, S. S. (2002) Biological consequences of
free radical-damaged DNA bases, Free Radic. Biol. Med.,
33, 1-14, doi: 10.1016/s0891-5849(02)00827-4.
2.Boiteux, S., and Guillet, M. (2004) Abasic sites in
DNA, repair and biological consequences in Saccharomyces
cerevisiae, DNA Repair, 3, 1-12, doi:
S1568786403002192.
3.Van Loon, B., Markkanen, E., and Hubscher, U.
(2010) Oxygen as a friend and enemy: how to combat the mutational
potential of 8-oxo-guanine, DNA Repair (Amst), 9,
604-616, doi: 10.1016/j.dnarep.2010.03.004.
4.Kuznetsova, A. A., Knorre, D. G., and Fedorova, O.
S. (2009) Oxidation of DNA and its components with reactive oxygen
species, Russ. Chem. Rev., 78, 659-678,
doi: 10.1070/Rc2009v078n07abeh004038.
5.Nakamura, J., Walker, V. E., Upton, P. B., Chiang,
S. Y., Kow, Y. W., and Swenberg, J. A. (1998) Highly sensitive
apurinic/apyrimidinic site assay can detect spontaneous and chemically
induced depurination under physiological conditions, Cancer
Res., 58, 222-225.
6.Frederico, L. A., Kunkel, T. A., and Shaw, B. R.
(1990) A sensitive genetic assay for the detection of cytosine
deamination, determination of rate constants and the activation energy,
Biochemistry, 29, 2532-2537.
7.Burcham, P. C. (1999) Internal hazards, baseline
DNA damage by endogenous products of normal metabolism, Mutat.
Res., 443, 11-36.
8.Evans, M. D., Dizdaroglu, M., and Cooke, M. S.
(2004) Oxidative DNA damage and disease, induction, repair and
significance, Mutat. Res., 567, 1-61.
9.Leandro, G. S., Sykora, P., and Bohr, V. A. (2015)
The impact of base excision DNA repair in age-related neurodegenerative
diseases, Mutat. Res. Mol. Mech. Mutagen., 776, 31-39,
doi: 10.1016/j.mrfmmm.2014.12.011.
10.Coppede, F., and Migliore, L. (2015) DNA damage
in neurodegenerative diseases, Mutat. Res. Mol. Mech. Mutagen.,
776, 84-97, doi: 10.1016/j.mrfmmm.2014.11.010.
11.Helleday, T., Petermann, E., Lundin, C., Hodgson,
B., and Sharma, R. A. (2008) DNA repair pathways as targets for cancer
therapy, Nat. Rev. Cancer, 8, 193-204, doi:
10.1038/nrc2342.
12.Dietlein, F., Thelen, L., and Reinhardt, H. C.
(2014) Cancer-specific defects in DNA repair pathways as targets for
personalized therapeutic approaches, Trends Genet., 30,
326-339, doi: 10.1016/j.tig.2014.06.003.
13.Trushina, E., and McMurray, C. T. (2007)
Oxidative stress and mitochondrial dysfunction in neurodegenerative
diseases, Neuroscience, 145, 1233-1248,
doi: 10.1016/j.neuroscience.2006.10.056.
14.Cooke, M. S., Evans, M. D., Dizdaroglu, M., and
Lunec, J. (2003) Oxidative DNA damage, mechanisms, mutation, and
disease, FASEB J., 17, 1195-1214.
15.Gros, L., Saparbaev, M. K., and Laval, J. (2002)
Enzymology of the repair of free radicals-induced DNA damage,
Oncogene, 21, 8905-8925.
16.Zharkov, D. O. (2008) Base excision DNA repair,
Cell. Mol. Life Sci., 65, 1544-1565,
doi: 10.1007/s00018-008-7543-2.
17.Klungland, A., Rosewell, I., Hollenbach, S.,
Larsen, E., Daly, G., Epe, B., Seeberg, E., Lindahl, T., and Barnes, D.
E. (1999) Accumulation of premutagenic DNA lesions in mice defective in
removal of oxidative base damage, Proc. Natl. Acad. Sci. USA,
96, 13300-13305.
18.Parsons, J. L., and Elder, R. H. (2003) DNA
N-glycosylase deficient mice, a tale of redundancy, Mutat. Res.,
531, 165-175.
19.Nilsen, H., Rosewell, I., Robins, P., Skjelbred,
C. F., Andersen, S., Slupphaug, G., Daly, G., Krokan, H. E., Lindahl,
T., and Barnes, D. E. (2000) Uracil-DNA glycosylase (UNG)-deficient
mice reveal a primary role of the enzyme during DNA replication,
Mol. Cell, 5, 1059-1065.
20.Fung, H., and Demple, B. (2005) A vital role for
Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells,
Mol. Cell, 17, 463-470,
doi: 10.1016/j.molcel.2004.12.029.
21.Xanthoudakis, S., Smeyne, R. J., Wallace, J. D.,
and Curran, T. (1996) The redox/DNA repair protein, Ref-1, is essential
for early embryonic development in mice, Proc. Natl. Acad. Sci.
USA, 93, 8919-8923, doi: 10.1073/Pnas.93.17.8919.
22.Unnikrishnan, A., Raffoul, J. J., Patel, H. V.,
Prychitko, T. M., Anyangwe, N., Meira, L. B., Friedberg, E. C.,
Cabelof, D. C., and Heydari, A. R. (2009) Oxidative stress alters base
excision repair pathway and increases apoptotic response in
apurinic/apyrimidinic endonuclease 1/redox factor-1 haploinsufficient
mice, Free Radic. Biol. Med., 46, 1488-1499,
doi: 10.1016/j.freeradbiomed.2009.02.021.
23.Gines, G., Saint-Pierre, C., and Gasparutto, D.
(2014) On-bead fluorescent DNA nanoprobes to analyze base excision
repair activities, Anal. Chim. Acta, 812, 168-175,
doi: 10.1016/j.aca.2013.12.038.
24.Chaim, I. A., Nagel, Z. D., Jordan, J. J.,
Mazzucato, P., Ngo, L. P., and Samson, L. D. (2017) In vivo
measurements of interindividual differences in DNA glycosylases and
APE1 activities, Proc. Natl. Acad. Sci. USA, 114,
e10379-e10388, doi: 10.1073/pnas.1712032114.
25.Maksimenko, A., Ishchenko, A. A., Sanz, G.,
Laval, J., Elder, R. H., and Saparbaev, M. K. (2004) A molecular beacon
assay for measuring base excision repair activities, Biochem.
Biophys. Res. Commun., 319, 240-246,
doi: 10.1016/j.bbrc.2004.04.179.
26.Lebedeva, N. A., Anarbaev, R. O., Kupryushkin, M.
S., Rechkunova, N. I., Pyshnyi, D. V., Stetsenko, D. A., and Lavrik, O.
I. (2015) Design of a new fluorescent oligonucleotide-based assay for a
highly specific real-time detection of apurinic/apyrimidinic site
cleavage by tyrosyl-DNA phosphodiesterase 1, Bioconjug. Chem.,
26, 2046-2053, doi: 10.1021/acs.bioconjchem.5b00451.
27.Thomson, G. J., Hamilton, N. S., Hopkins, G. V.,
Waddell, I. D., Watson, A. J., and Ogilvie, D. J. (2013) A
fluorescence-based assay for the apurinic/apyrimidinic-site cleavage
activity of human tyrosyl-DNA phosphodiesterase 1, Anal.
Biochem., 440, 1-5, doi: 10.1016/j.ab.2013.05.003.
28.Li, J., Svilar, D., McClellan, S., Kim, J.-H.,
Ahn, E. E., Vens, C., Wilson, D. M., and Sobol, R. W. (2018) DNA repair
molecular beacon assay, a platform for real-time functional analysis of
cellular DNA repair capacity, Oncotarget, 9,
31719-31743, doi: 10.18632/oncotarget.25859.
29.Wilson, D. L., and Kool, E. T. (2018) Fluorescent
probes of DNA repair, ACS Chem. Biol., 13, 1721-1733,
doi: 10.1021/acschembio.7b00919.
30.Hu, J., Liu, M.-H., Li, Y., Tang, B., and Zhang,
C.-Y. (2018) Simultaneous sensitive detection of multiple DNA
glycosylases from lung cancer cells at the single-molecule level,
Chem. Sci., 9, 712-720, doi: 10.1039/c7sc04296e.
31.Svilar, D., Vens, C., and Sobol, R. W. (2012)
Quantitative, real-time analysis of base excision repair activity in
cell lysates utilizing lesion-specific molecular beacons, J. Vis.
Exp., 66, e4168, doi: 10.3791/4168.
32.Healing, E., Charlier, C. F., Meira, L. B., and
Elliott, R. M. (2019) A panel of colorimetric assays to measure
enzymatic activity in the base excision DNA repair pathway, Nucleic
Acids Res., 47, e61, doi: 10.1093/nar/gkz171.
33.Zhang, Y., Li, C., Tang, B., and Zhang, C. (2017)
Homogeneously sensitive detection of multiple DNA glycosylases with
intrinsically fluorescent nucleotides, Anal. Chem., 89,
7684-7692, doi: 10.1021/acs.analchem.7b01655.
34.Liu, G., He, W., and Liu, C. (2019) Sensitive
detection of uracil-DNA glycosylase (UDG) activity based on terminal
deoxynucleotidyl transferase-assisted formation of fluorescent copper
nanoclusters (CuNCs), Talanta, 195, 320-326,
doi: 10.1016/j.talanta.2018.11.083.
35.Du, Y.-C., Jiang, H.-X., Huo, Y.-F., Han, G.-M.,
and Kong, D.-M. (2016) Optimization of strand displacement
amplification-sensitized G-quadruplex DNAzyme-based sensing system and
its application in activity detection of uracil-DNA glycosylase,
Biosens. Bioelectron., 77, 971-977,
doi: 10.1016/j.bios.2015.10.080.
36.Zhang, L., Zhao, J., Jiang, J., and Yu, R. (2012)
A target-activated autocatalytic DNAzyme amplification strategy for the
assay of base excision repair enzyme activity, Chem. Commun.,
48, 8820-8822, doi: 10.1039/c2cc34531e.
37.Wang, L., Ren, M., Zhang, Q., Tang, B., and
Zhang, C. (2017) Excision repair-initiated enzyme-assisted bicyclic
cascade signal amplification for ultrasensitive detection of uracil-DNA
glycosylase, Anal. Chem., 89, 4488-4494,
doi: 10.1021/acs.analchem.6b04673.
38.Kladova, O. A., Bazlekowa-Karaban, M., Baconnais,
S., Pietrement, O., Ishchenko, A. A., Matkarimov, B. T., Iakovlev, D.
A., Vasenko, A., Fedorova, O. S., Le Cam, E., Tudek, B., Kuznetsov, N.
A., and Saparbaev, M. (2018) The role of the N-terminal domain
of human apurinic/apyrimidinic endonuclease 1, APE1, in DNA glycosylase
stimulation, DNA Repair (Amst), 64, 10-25,
doi: 10.1016/j.dnarep.2018.02.001.
39.Kuznetsova, A. A., Kuznetsov, N. A., Ishchenko,
A. A., Saparbaev, M. K., and Fedorova, O. S. (2014) Pre-steady-state
fluorescence analysis of damaged DNA transfer from human DNA
glycosylases to AP-endonuclease APE1, Biochim. Biophys. Acta,
1840, 3042-3051, doi: 10.1016/j.bbagen.2014.07.016.
40.Waters, T. R., Gallinari, P., Jiricny, J., and
Swann, P. F. (1999) Human thymine DNA glycosylase binds to apurinic
sites in DNA but is displaced by human apurinic endonuclease 1, J.
Biol. Chem., 274, 67-74.
41.Xia, L., Zheng, L., Lee, H. W., Bates, S. E.,
Federico, L., Shen, B., and O’Connor, T. R. (2005) Human
3-methyladenine-DNA glycosylase. Effect of sequence context on
excision, association with PCNA, and stimulation by AP-endonuclease,
J. Mol. Biol., 346, 1259-1274,
doi: 10.1016/j.jmb.2005.01.014.
42.Esadze, A., Rodriguez, G., Cravens, S. L., and
Stivers, J. T. (2017) AP-endonuclease 1 accelerates turnover of human
8-oxoguanine DNA glycosylase by preventing retrograde binding to the
abasic-site product, Biochemistry, 56, 1974-1986,
doi: 10.1021/acs.biochem.7b00017.
43.Baldwin, M. R., and O’Brien, P. J. (2009)
Human AP-endonuclease 1 stimulates multiple-turnover base excision by
alkyladenine DNA glycosylase, Biochemistry, 48,
6022-6033, doi: 10.1021/bi900517y.
44.Miroshnikova, A. D., Kuznetsova, A. A., Vorobjev,
Y. N., Kuznetsov, N. A., and Fedorova, O. S. (2016) Effects of mono-
and divalent metal ions on DNA binding and catalysis of human
apurinic/apyrimidinic endonuclease 1, Mol. BioSyst., 12,
1527-1539, doi: 10.1039/c6mb00128a.
45.Kuznetsov, N. A., Koval, V. V., and Fedorova, O.
S. (2011) Mechanism of recognition and repair of damaged DNA by human
8-oxoguanine DNA glycosylase hOGG1, Biochemistry (Moscow),
76, 118-130, doi: 10.1134/S0006297911010123.
46.Kladova, O. A., Kuznetsova, A. A., Fedorova, O.
S., and Kuznetsov, N. A. (2017) Mutational and kinetic analysis of
lesion recognition by Escherichia coli endonuclease VIII,
Genes, 8, 1-13, doi: 10.3390/genes8050140.
47.Kuznetsova, A. A., Iakovlev, D. A., Misovets, I.
V., Ishchenko, A. A., Saparbaev, M. K., Kuznetsov, N. A., and Fedorova,
O. S. (2017) Pre-steady-state kinetic analysis of damage recognition by
human single-strand selective monofunctional uracil-DNA glycosylase
SMUG1, Mol. BioSyst., 13, 2638-2649,
doi: 10.1039/c7mb00457e.
48.Yakovlev, D. A., Kuznetsova, A. A., Fedorova, O.
S., and Kuznetsov, N. A. (2017) Search for modified DNA sites with the
human methyl-CpG-binding enzyme MBD4, Acta Naturae, 9,
88-98.