REVIEW: Manipulating Cellular Energetics to Slow Aging of Tissues and Organs
S. S. Sokolov1 and F. F. Severin1,a*
1Lomonosov Moscow State University, Belozersky Institute of Physico-Chemical Biology, 119991 Moscow, Russia* To whom correspondence should be addressed.
Received April 11, 2020; Revised April 30, 2020; Accepted May 1, 2020
Up to now numerous studies in the field of gerontology have been published. Nevertheless, a well-known food restriction remains the most reliable and efficient way of lifespan extension. Physical activity is also a well-documented anti-aging intervention being especially efficient in slowing down the age-associated decline of skeletal muscle mass. In this review we focus on the molecular mechanisms of the effect of physical exercise on muscle tissues. We also discuss the possibilities of pharmacological extension of this effect to the rest of the tissues. During the exercise, the level of ATP decreases triggering activation of AMP-dependent protein kinase (AMPK). This kinase stimulates antioxidant potential of the cells and their mitochondrial respiratory capacity. The exercise also induces mild oxidative stress, which, in turn, mediates the stimulation via hormetic response. Furthermore, during the exercise cells generate activators of mammalian target of rapamycin (mTOR). The intracellular ATP level increases during the rest periods between exercises thus promoting mTOR activation. Therefore, regular exercise intermittently activates anti-oxidant defenses and mitochondrial biogenesis (via AMPK and the hormetic response) of the muscle tissue, as well as its proliferative potential (via mTOR), which, in turn, impedes the age-dependent muscle atrophy. Thus, the intermittent treatment with activators of (i) AMPK combined with the inducers of hormetic response and of (ii) mTOR might partly mimic the effects of physical exercise. Importantly, pharmacological activation of AMPK takes place in the absence of ATP level decrease. The use of uncouplers of respiration and oxidative phosphorylation at the phase of AMPK activation could also prevent negative consequences of the cellular hyper-energization. It is believed that the decline of both antioxidant and proliferative potentials of the cells causes the age-dependent decline of multiple tissues, rather than only the muscular one. We argue that the approach above is applicable for the majority of tissues in an organism.
KEY WORDS: aging, caloric restriction, uncouplers, geroprotectors, mTOR, AMPKDOI: 10.1134/S0006297920060024
Abbreviations: AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; AMPK, AMP-dependent protein kinase; GH, growth hormone; IGF1, insulin-like growth factor 1; MGF, muscle growth factor; mTOR, mammalian target of rapamycin; ROS, reactive oxygen species.
INTRODUCTION
Over the last few decades, we have witnessed a significant breakthrough in the field of cell biology. Currently the key mechanisms of cell division, differentiation, stress response, senescence, and programmed death are most likely have been elucidated. At the same time, the progress in understanding the biology of a single cell did not lead to any major achievements in the field of aging. Therefore, one could expect that the coming advances in understanding of the aging process will take place at the level above the cellular one, i.e., at the level of tissues and organs.
The lifespan of experimental animals, mice and rats, is typically increased by pharmacological substances only by 10-30%. The most well studied life-extending chemicals are rapamycin, salicylate, resveratrol, metformin, 2-deoxyglucose, spermidine, and several others (see [1] for review). Food restriction, which has been known literally for ages as intervention improving healthspan and lifespan, shows approximately the same effect as the pharmacological treatment (see [2-6] for review).
Physical activity is also well-known for its anti-aging effects. For example, a possibility to run in a wheel leads to approximately 10% lifespan extension in rats [7]. In comparison with food restriction, physical exercise in the majority of mammalian model systems produces health span rather than lifespan extension (reviewed in [2]). In the case of elderly people, positive effect of physical activity is mostly due to preservation of the muscle functionality [8]. Indeed, a lot of studies have shown physical exercise to be especially efficient in preventing age-dependent degeneration of skeletal muscles [9]. In particular, it was reported that by the age of 20 months the grip strength in the control group of rats displayed approximately a two-fold decrease [10]. At the same time, only 10% loss of the grip strength was observed in the group of rats subjected to a long-term physical exercise (see Fig. 3 in this paper). In addition, it was shown that the running time until exhaustion and the maximum running speed were twice lower in the group of 26-month-old control rats in comparison with the 8-month-old ones [11]. On the contrary, the rats subjected to chronic physical exercise did not show such age-dependent decline ([11], Fig. 3).
Therefore, one can conclude that the effect of food restriction on lifespan extension (approximately 30%) is much lower than that of chronic physical exercise on age-dependent muscle degeneration (several fold). It is important to note that the molecular mechanisms underlying positive impact of physical exercise on skeletal muscle are well studied. These involve basic mechanisms of activation of antioxidant systems, improvement of general physiology, and prevention of the tissue atrophy (reviewed in the next section). Pharmacological stimulation of these mechanisms, obviously, could be beneficial not only for muscle but for many other tissues. Indeed, a number of pharmacological substances called exercise mimetics (such as metformin, resveratrol) were shown to extend the lifespans of a set of model organisms [12, 13].
Since physical activity triggers not one but several intracellular signalling cascades, none of the currently known exercise mimetic substances used individually can fully reproduce the effect of physical activity on skeletal muscle. Moreover, the main signalling cascades activated by physical activity, AMPK-dependent and mTOR-dependent ones, inhibit each other. Therefore, temporal alternating of the stimulation is necessary. Interestingly enough, intermittent fasting was shown to be no less effective than the usual caloric restriction in terms of lowering blood glucose and cholesterol levels [14, 15].
Pharmacological substances activating AMP-dependent protein kinase (AMPK) and mammalian target of rapamycin (mTOR) do exist, some of them are classified as performance-enhancing drugs in sports. The reason for the negative effects of mTOR stimulants is obvious. Being an antagonist of AMPK, hyper-activated mTOR switches cell metabolism from respiration to glycolysis, and provokes inflammatory processes. AMPK stimulants appear to be less dangerous. Most of them are antidiabetic drugs. Despite the positive effect on muscle tissue, muscle atrophy was observed with prolonged use of 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), the most popular of these drugs (reviewed in [16]). One of the reasons for the side effects is probably that activation of AMPK under conditions of high levels of nutrients leads to abnormal increase in the level of intracellular ATP. This can cause hyperpolarization of mitochondria, resulting in the generation of ROS in the respiratory chain. An abnormal increase in ATP level and, as a result, depletion of ADP can have other negative consequences. Recently we have hypothesized that a moderate, controlled decrease in the level of ATP and the membrane potential of mitochondria can be achieved by using substances that uncouple mitochondrial respiration and ATP synthesis [17].
In this review, we have attempted to suggest combinations of the pharmacological preparations and dietary supplements that can best mimic the effect of physical activity on skeletal muscles, and thus expand this effect to the most of other tissues. In the following section we summarize the molecular mechanisms underlying the effects of physical activity on muscle tissues.
MOLECULAR MECHANISMS OF THE ANTIAGING EFFECT OF PHYSICAL ACTIVITY
ON SKELETAL MUSCLES
One of the main ways of age-dependent degeneration of skeletal muscles is sarcopenia, a process initiated by the disappearance of neuromuscular junctions. It is believed that such denervation of muscle fibers is due to the problems with homeostasis of calcium, the key signaling molecule in the degeneration of myofibrils. Apparently, the main reason for the disruptions of Ca2+ trafficking in the muscle cell is its leakage from sarcoplasmic reticulum caused by oxidative stress [18]. In addition, skeletal muscle proliferation decreases with age (see above). In this section, we argue that chronic physical exercise increases both the antioxidant and the proliferative potential of skeletal muscles.
Hormetic response to mild oxidative stress. Physical exercise causes a mild oxidative stress in skeletal muscles. The main sources of this stress are production of hydrogen peroxide by NADPH oxidase Nox2 (reviewed in [19]), as well as the generation of mitochondrial reactive oxygen species, ROS [20]. It is known that this oxidative stress induces a compensatory, hormetic effect by activating redox-regulated transcription factors such as PPARγ, Nrf2, HIF-1, NF-κB [21-23]. Activation of the transcription factors leads directly to the increase in the cell resistance to oxidative stress by stimulating the expression of antioxidant enzymes. In addition, there are indirect antioxidant effects caused by activation of transcription, such as acceleration of mitochondrial biogenesis and changes in calcium homeostasis in the muscle cell [23].
AMPK activation. During exercise ATP hydrolysis is activated in muscle cells, ADP concentration increases and, as a result of 2ADP⇄ATP + AMP reaction, AMP-dependent protein kinase is activated [24-27]. Furthermore, the NAD+/NADH ratio increases thus leading to activation of Sirt1 deacetylase. This enzyme additionally activates AMPK and also increases the resistance of the cells to oxidative stress (reviewed in [28], [17]). Subsequently, the antioxidant potential of the cell increases, mitochondrial biogenesis is activated.
Interestingly enough, AMPK is also activated during skeletal muscle degeneration induced by the prolonged absence of physical activity. Several studies have shown that the absence of muscle contractions leads to muscle atrophy. This atrophy appears to be due to mitochondrial dysfunction, which leads to activation of AMPK [29]. This fact is in agreement with the observation that activation of AMPK can lead to cell death [30-32]. In particular, it has been recently shown that HSF1, a key transcription factor stimulating cell survival and proliferation under stressful conditions, and AMPK inhibit each other [33]. It is likely that the physiological role of AMPK-mediated atrophy of unused muscles is to get rid of unnecessary cells under the conditions of energy deficiency.
Activation of mTOR. The generation of activators of mTOR, the AMPK antagonist signaling complex, is well known to take place during exercise. Exercise stimulates mTOR of a working muscle in at least three ways. First, stretching of Z-disk triggers intracellular activation cascade. Second, actively working muscle secrets Muscle Growth Factor (MGF), an endocrine activator of mTOR. MGF is a splice variant of the well-known insulin-like growth factor (IGF1). IGF1 is one of the key anabolic hormones stimulating proliferation. Third, in response to intense muscle function, hypothalamus secretes growth hormone (GH), which is also a central endocrine activator of mTOR in most of the cell types, including muscle tissue [34].
It is important to note that physical activity is both an activator and an inhibitor of skeletal muscle growth. As mentioned above, muscle activity leads to an increase of the ADP/ATP ratio and, as a consequence, activation of AMPK. Being an antagonist of mTOR, during exercise AMPK inhibits mTOR, and this inhibitory effect is stronger than that of the activating one. Upon transition into the resting state, intracellular ATP levels rise, which allows mTOR activation [35].
Myokine secretion. In addition to MGF, physical activity triggers muscular secretion of a number of signaling molecules called myokines. The list of myokines includes a number of interleukins, irisin, meteorin, FGF-21 (Fibroblast Growth Factor), and GDF-15 (Growth and Differentiation Factor). It is believed that myokines contribute to mobilization of the energy resources and suppress inflammatory processes under the conditions of increased energy consumption due to intense muscular activity. Myokine receptors are found in many tissues: myokines positively affect physiology of the body, their effect is similar to the one of caloric restriction [36-38]. Within the scope of this section, it should be mentioned that at least one of the myokines, GDF-15, also affects muscle tissue directly. As in the case of AMPK activation, the effect of GDF-15 on muscles is dual: this myokine improves stress resistance of muscle tissue [39, 40], but also can cause its atrophy [38, 41, 42].
SIMULATION OF PHYSICAL ACTIVITY: DIET AND PHARMACEUTICALS
Is it possible to simulate the effect of physical exercise by activating the abovementioned signaling pathways pharmacologically? This issue is especially important because stimulation by physical activity is not possible for most non-muscle tissues. Indeed, following our logics, alternate administration of AMPK and mTOR stimulants can theoretically affect most of the tissues in a way physical activity affects skeletal muscle. It is worth noting here that an increasing number of works on intermittent fasting is being published. In particular, it was shown that the mice that for a year were allowed free access to food only twice a day, one hour in the morning and one hour in the evening, significantly improved their physiological parameters. In particular, plasma glucose and triglycerides levels decreased, while sensitivity to insulin increased. In contrast to the usual caloric restriction, intermittent fasting did not lead to the decrease in muscle mass [14]. Similar results were obtained in humans. For four days, a group of overweight people ate only from eight in the morning until two in the afternoon. Significant improvement in the metabolism of glucose and lipids was observed following such a short period of time [15].
At the same time, it seems unlikely that such intermittent fasting is a significantly more effective way to delay aging than the well-established caloric restriction. Otherwise, over the course of many thousands of years of the history of mankind, such fasting regime would most likely have been discovered empirically and extensively used. Below we present possible approaches to more effective stimulation of both mTOR and AMPK.
mTOR stimulation. At present, a large number of mTOR activator substances are known. Many of them are classified as prohibited performance-enhancing drugs in sports due to their side effects. One of the reasons for the negative consequences of the use of mTOR stimulants (anabolics: growth hormone, IGF1, testosterone, oxymetalon) is obvious. Being a Sirt1/AMPK antagonist, hyper-activated mTOR switches cell metabolism from respiration to glycolysis, and provokes inflammatory processes. The pharmacological inhibitor of mTOR, rapamycin, prolongs the life of experimental animals; its effect is comparable to that of food restrictions [2-4]. For this reason, the treatments not increasing but decreasing the level of GH are mainly considered to be anti-aging ones (see review [43]). Nevertheless, there have been several attempts to use GH and its agonists to delay aging. Indeed, in the old age, GH concentration in human blood is significantly reduced. This was the main rationale behind treating the elderly with IGF1 or GH. In particular, positive effect on the muscle mass and general well-being of patients was observed [43-45].
All these facts are consistent with the main hypothesis of this review on the benefits of alternating stimulation of mTOR and AMPK. What is the best way to stimulate mTOR? On the one hand, unlimited, plentiful nutrition is not the most effective way to activate mTOR. Indeed, combining an appropriate diet with physical activity and/or anabolic substances is a much more effective way to increase muscle mass than the diet alone. On the other hand, it seems intuitively clear that the use of anabolics (performance-enhancing drugs in sports) is hardly compatible with the concept of delaying aging. Stimulation of mTOR with the help of dietary supplements seems to be an intermediate approach. It is well known that mTOR is a sensor of the concentration of amino acids in a cell; therefore, ordinary amino acids are activators of mTOR. Branched side chain amino acids are most effective in terms of mTOR activation [46]. In particular, in order to combat sarcopenia and to increase the muscle mass of athletes, leucine supplements were used. As it turned out, the metabolite of leucine, hydroxymethyl butyrate (HMB), as well as L-citrulline (reviews [47-49]) are more effective in this respect. To the best of our knowledge, these substances have never been used in the experiments on slowing down aging of mammals. According to the logics of this review, they can be used for this purpose in combination with other exercise mimetics.
AMPK stimulation. AMPK activators appear to be less hazardous for health than mTOR stimulants, anabolics. Some of them (metformin, resveratrol) are well known anti-diabetic drugs. Even AICAR, being classified as a performance-enhancing drug, is used to treat diabetes. For the short-term use, they, like physical exercise, improve general physiology of muscles and, in particular, functionality of mitochondria. At the same time, the prolonged use of AICAR resulted in the decrease of muscle mass due to chronic activation of catabolic processes [16]. Based on the abovementioned arguments, this negative side effect could be neutralized by periodic activation of mTOR. Are there any other arguments against the long-term use of AMPK activators?
We suggested previously that pharmacological stimulation of AMPK can cause hyper-energization of cells [17]. Indeed, active AMPK in the absence of nutrient deficiency can lead to abnormal increase in the level of cellular ATP. This can, in turn, lead to hyperpolarization of mitochondria, causing generation of ROS in the mitochondrial respiratory chain [17]. We have recently shown that ATP hydrolysis localized at the mitochondrial outer membrane due to mitochondrially-bound hexokinase activity is sufficient to prevent oxidative damage by mitochondrial ROS. We also demonstrated for a number of mammalian species that the activity of mitochondrial hexokinase positively correlates with their lifespan [50].
The abnormal increase in the level of ATP and, as a consequence, depletion of ADP could possibly have other unpredictable negative consequences associated, in particular, with glycolysis. Since the reversible reaction catalyzed by pyruvate kinase is associated with the synthesis of ATP from ADP, hyper-energization of the cell could lead to accumulation of the intermediate products of glycolysis. These products (triose phosphates, fructose-1,6-bisphosphate) are regulatory molecules. Specifically, these intermediates trigger the Crabtree effect: suppression of the energy function of mitochondria and stimulation of glycolysis [51].
Another potential problem that could arise upon hyper-energization of the cell is the decrease of the NAD+/NADH ratio. Indeed, glycolysis activation caused by AMPK stimulation should increase the rate of NADH synthesis, and high transmembrane potential of mitochondria should decrease the rate of its oxidation to NAD+ [17]. NAD+ is the rate-limiting substrate of deacetylases. Many studies have shown that these enzymes, the most known of which is histone deacetylase Sirt1, are involved in the regulation of longevity (see reviews [52, 53]). Perhaps, for this reason, we have failed to find the works in which the direct pharmacological activators of AMPK (AICAR, GW501516, MK-8722) increase the lifespan of model organisms.
Following these logics, it could be suggested that the optimal way to activate AMPK from the point of view of slowing aging is to increase the ADP/ATP ratio in cells. This is exactly what happens in skeletal muscle subjected to physical activity. Is it possible to achieve a mild, controlled decrease in ATP levels in all body tissues? The problem is that the level of blood glucose, which is the main energy currency of the body, is regulated very strictly. Indeed, under standard dietary restrictions used to prolong life, the blood glucose concentration in experimental mice and rats decreases by only 10-30% compared to that in animals with unlimited access to food [54, 55]. Such a decrease is obviously sufficient for activating the hormonal response in the special sensor tissues, which, in turn activate Sirt1/AMPK in the other tissues by secreting various signaling molecules (e.g., lipokines) (see review [17]). At the same time, energy consumption by a cell at rest and in an active state can differ several times. For example, activation of macrophages leads to approximately four-fold increase in glucose consumption [56]. It is also obvious that the standard caloric restriction is not able to lower the level of nutrients in many types of cells, which are in a direct contact with blood plasma. Let us consider, for example, endothelium of blood vessel capillaries. Almost all glucose entering the body diffuses through the GLUT1 transporter of the plasma membrane of the endothelial monolayer [57]. Thus, access to glucose in this type of cell is immeasurably higher than in the peripheral tissues.
Therefore, both in the case of stimulation using direct pharmacological activators and in the case of nutritional restriction, activation of AMPK in most tissues does not happen due to increase in the ratio of ADP/ATP in the cells. At the same time, it seems rather unlikely that artificial lowering blood glucose concentration in human body by several fold could increase life expectancy. The alternative way is pharmacological activation of AMPK in combination with the substances that reduce negative effects of cell hyper-energization.
Recently we presented a literature review on how a controlled decrease in ATP levels and mitochondrial membrane potential using uncouplers of respiration and oxidative phosphorylation can be used to slow aging [17]. The controlled decrease of the membrane potential could have multiple positive consequences for cell physiology, such as, for example, an enhanced control of mitochondria quality (see review [58]). It follows from the logics of our review that stimulation of AMPK by uncoupling can be even more effective than the usual nutritional restriction, since uncoupling can lower membrane potential and ATP level in all tissues. The attempt to slow the aging process in mice using the most well studied uncoupler, DNP (2,4-dinitrophenol) was described in the literature. Similar to the classic dietary restrictions, administration of DNP to mice via drinking water resulted in an approximately 30% decrease in blood glucose concentration and a moderate, less than 10%, increase in the life span of animals [59]. Perhaps, the relatively low efficiency of DNP as an anti-aging compound was due to insufficient degree of uncoupling. Indeed, the authors of the article used concentration of the substance corresponding to the consumption of less than one micromole per kilogram of animal weight. At the same time, the acting uncoupling concentration of DNP in cell culture has been reported to be 100 µmol/liter [60], and in a short-term experiment with mice it was 5.5 mmol/kg of the mouse weight [61]. However, even small doses of DNP protect neurons in a mouse model of Parkinson’s disease [62, 63]. Summarizing this part, it can be suggested that either DNP or other uncoupling agents (see review [17]) could potentially be used in combination with pharmacological activators to stimulate AMPK.
Imitation of the effects of myokines and of hermetic response to oxidative stress. As was mentioned above, the main muscle endorphins acting on the muscle itself are MGF and GDF-15. MGF is an activator of mTOR. Its effect can theoretically be imitated by biologically active food supplements (see above). Possibly, the easiest and safest way to activate GDF-15 secretion in the body is to use metformin. It has been shown recently that the effects of metformin as a mimetic of food restriction are largely due to the secretion of GDF-15 caused by its administration [64]. At the same time, GDF-15 is well known to be secreted by many organs in response to oxidative stress (see review [65]). Is metformin indeed an inducer of hormetic oxidative stress response? It has been shown that metformin inhibits complex I of mitochondrial respiratory chain (see review [66]). It is also well-documented that inhibition of this complex can cause superoxide generation [67-69].
The use of metformin is unlikely to be the only possible way to induce a mild oxidative stress. For example, vitamin K3, menadione, and its derivatives are widely used for such purposes. Notably, menadione was shown to increase the lifespan of C. elegans worms [70] and baker’s yeast [71] due to the hormetic effect. To our best knowledge, menadione was not used as an anti-aging compound in higher model organisms. At the same time, menadione and its analogues are used as anti-cancer agents because of their pro-oxidative action [72].
It should be noted that the use of antioxidants to slow down aging does not agree with the concept of the benefits of hermetic response to oxidative stress. In fact, there is practically no description in the literature of the successful examples of prolonging the life of mammals using antioxidants. Moreover, the use of standard low molecular weight antioxidants can increase the risk of cancer (see reviews [73, 74]).
The exception, obviously, is the mitochondria-targeted antioxidant SkQ1. There have been several successful attempts both to prolong the life of mice and to cure the number of age-dependent diseases using SkQ1 (see reviews [73, 75-77]). Perhaps the reason for being an exception is that SkQ1 decreases ROS level locally, in mitochondria, but not in cytoplasm. It is known that it is precisely cytoplasmic hydrogen peroxide that reacts with various transcription factors and, in this way, causes a hormetic response [78, 79]. In addition, it was recently shown that plastoquinone, the antioxidant part of SkQ1 molecule, could react with superoxide. As a result of this reaction, superoxide is transformed into hydrogen peroxide: PQH2 + O2.– = PQ.– + H2O2 [80, 81]. According to the authors, equilibrium constant of this reaction is higher than 109 [81]. It follows from these data that, although SkQ1 is an antioxidant (protection against superoxide), it can simultaneously activate the cascade of cell protection against ROS (generation of hydrogen peroxide, which diffuses easily through the membranes). Presumably, the combination of pro-oxidant with SkQ1 might initiate a hormetic response to oxidative stress.
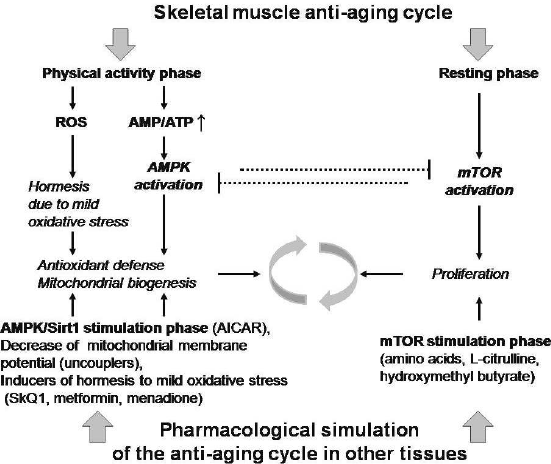
Summing up, we suggest that alternating pharmacological stimulation of mTOR and AMPK can be effective in prolonging functionality of the most of the tissues. In addition, we speculate that the use of uncoupling at the stage of AMPK stimulation can suppress negative effects associated with the cell hyper-energization. In parallel with the stimulation of AMPK, we propose to simulate the hormetic response to oxidative stress. The figure highlights the effects of physical exercise on skeletal muscles mentioned in this review and the proposed methods for their imitation in non-muscle tissues. We emphasize again that the effects of mTOR activation, on the one hand, and of other stimuli listed in the figure, on the other hand, are oppositely directed and inhibit each other. Therefore, similar to intermittent fasting, in order to achieve the desired effect, alternation of two types of effects seems to be necessary. The durations of alternating periods will obviously depend on pharmacokinetics of the specific substances.
It is believed that a decrease of both antioxidant and proliferative potential of cells is the cause of aging of many tissues, not only the muscle ones (see review [82]). From our point of view, the above anti-aging strategy can be applied to most types of cells in the human body. Obviously, it is impossible to predict how the proposed set of stimulants would affect each of the tissues. At the same time, it is rather obvious that the age-dependent degeneration of various tissues affects the aging of the body to different degrees. Which of the tissues limits the life span of a human being? Statistic indicates four main causes of death in humans: cancer, diabetes, cardiovascular diseases, and chronic obstructive pulmonary disease (see reviews [83, 84]). Therefore, it can be suggested that human lifespan is limited by the immune system, glucose sensor tissues, and also endothelial tissues of blood vessels and lungs. From our point of view, the proposed set of stimuli is at least not harmful for each of these tissues.
Mechanisms of anti-aging effects of periodic physical exercise on muscle tissue and expected effects of pharmacological mimetics of physical exercise on non-muscle tissues.
Funding. This work was supported by the Russian Foundation for Basic Research (project No. 19-14-50642).
Conflict of interest. The authors declare no conflict of interest.
Ethical approval. This article does not contain description of studies involving humans or animals as research subjects performed by any of the authors.
REFERENCES
1.Qian, M., and Liu, B. (2018) Pharmaceutical
intervention of aging, Adv. Exp. Med. Biol., 1086,
235-254, doi: 10.1007/978-981-13-1117-8_15.
2.Mercken, E. M., Carboneau, B. A., Krzysik-Walker,
S. M., and de Cabo, R. (2012) Of mice and men: the benefits of caloric
restriction, exercise, and mimetics, Ageing Res. Rev.,
11, 390-398, doi: 10.1016/j.arr.2011.11.005.
3.Palliyaguru, D. L., Moats, J. M., Di Germanio, C.,
Bernier, M., and de Cabo, R. (2019) Frailty index as a biomarker of
lifespan and healthspan: focus on pharmacological interventions,
Mech. Ageing Dev., 180, 42-38,
doi: 10.1016/j.mad.2019.03.005.
4.Martel, J., Ojcius, D. M., Ko, Y.-F., Chang, C.-J.,
and Young, J. D. (2019) Antiaging effects of bioactive molecules
isolated from plants and fungi, Med. Res. Rev., 39,
1515-1552, doi: 10.1002/med.21559.
5.Fontana, L., Partridge, L., and Longo, V. D. (2010)
Extending healthy life span – from yeast to humans,
Science, 328, 321-326, doi: 10.1126/science.1172539.
6.Mulvey, L., Sinclair, A., and Selman, C. (2014)
Lifespan modulation in mice and the confounding effects of genetic
background, J. Genet. Genomics, 41, 497-503,
doi: 10.1016/j.jgg.2014.06.002.
7.Holloszy, J. O. (1998) Longevity of exercising male
rats: effect of an antioxidant supplemented diet, Mech. Ageing
Dev., 100, 211-219,
doi: 10.1016/s0047-6374(97)00140-1.
8.Prado, C. M., Purcell, S. A., Alish, C., Pereira,
S. L., Deutz, N. E., Heyland, D. K., Goodpaster, B. H., Tappenden, K.
A., and Heymsfield, S. B. (2018) Implications of low muscle mass across
the continuum of care: a narrative review, Ann. Med., 50,
675-693, doi: 10.1080/07853890.2018.1511918.
9.Distefano, G., and Goodpaster, B. H. (2018) Effects
of exercise and aging on skeletal muscle, Cold Spring Harb.
Perspect. Med., 8,
doi: 10.1101/cshperspect.a029785.
10.Hernández-Álvarez, D., Mena-Montes,
B., Toledo-Pérez, R., Pedraza-Vázquez, G.,
López-Cervantes, S. P., Morales-Salazar, A.,
Hernández-Cruz, E., Lazzarini-Lechuga, R.,
Vázquez-Cárdenas, R. R., Vilchis-DeLaRosa, S.,
Posadas-Rodríguez, P., Santín-Márquez, R.,
Rosas-Carrasco, O., **Ibañez-Contreras, A.,
Alarcón-Aguilar, A., López-Díazguerrero, N. E.,
Luna-López, A., and Königsberg, M. (2019) Long-term
moderate exercise combined with metformin treatment induces an hormetic
response that prevents strength and muscle mass loss in old female
wistar rats, Oxid. Med. Cell. Longev., 2019, 3428543,
doi: 10.1155/2019/3428543.
11.Li, F.-H., Sun, L., Zhu, M., Li, T., Gao, H.-E.,
Wu, D.-S., Zhu, L., Duan, R., and Liu, T. C. (2018) Beneficial
alterations in body composition, physical performance, oxidative
stress, inflammatory markers, and adipocytokines induced by long-term
high-intensity interval training in an aged rat model, Exp.
Gerontol., 113, 150-162,
doi: 10.1016/j.exger.2018.10.006.
12.Klimova, B., Novotny, M., and Kuca, K. (2018)
Anti-aging drugs – prospect of longer life? Curr. Med.
Chem., 25, 1946-1953,
doi: 10.2174/0929867325666171129215251.
13.Carmona, J. J., and Michan, S. (2016) Biology of
healthy aging and longevity, Rev. Invest. Clin., 68,
7-16.
14.Martinez-Lopez, N., Tarabra, E., Toledo, M.,
Garcia-Macia, M., Sahu, S., Coletto, L., Batista-Gonzalez, A.,
Barzilai, N., Pessin, J. E., Schwartz, G. J., Kersten, S., and Singh,
R. (2017) System-wide benefits of intermeal fasting by autophagy,
Cell Metab., 26, 856-871,
doi: 10.1016/j.cmet.2017.09.020.
15.Jamshed, H., Beyl, R. A., Della Manna, D. L.,
Yang, E. S., Ravussin, E., and Peterson, C. M. (2019) Early
time-restricted feeding improves 24-hour glucose levels and affects
markers of the circadian clock, aging, and autophagy in humans,
Nutrients, 11, doi: 10.3390/nu11061234.
16.Hawley, J. A., and Holloszy, J. O. (2009)
Exercise: it’s the real thing! Nutr. Rev., 67,
172-178, doi: 10.1111/j.1753-4887.2009.00185.x.
17.Knorre, D. A., and Severin, F. F. (2016)
Uncouplers of oxidation and phosphorylation as antiaging compounds,
Biochemistry (Moscow), 81, 1438-1444,
doi: 10.1134/S0006297916120051.
18.Espinosa, A., Henríquez-Olguín, C.,
and Jaimovich, E. (2016) Reactive oxygen species and calcium signals in
skeletal muscle: a crosstalk involved in both normal signaling and
disease, Cell Calcium, 60, 172-179,
doi: 10.1016/j.ceca.2016.02.010.
19.Ferreira, L. F., and Laitano, O. (2016)
Regulation of NADPH oxidases in skeletal muscle, Free Radic. Biol.
Med., 98, 18-28,
doi: 10.1016/j.freeradbiomed.2016.05.011.
20.Ji, L. L., Kang, C., and Zhang, Y. (2016)
Exercise-induced hormesis and skeletal muscle health, Free Radic.
Biol. Med., 98, 113-22,
doi: 10.1016/j.freeradbiomed.2016.02.025.
21.Musci, R. V., Hamilton, K. L., and Linden, M. A.
(2019) Exercise-induced mitohormesis for the maintenance of skeletal
muscle and healthspan extension, Sports (Basel), 7,
doi: 10.3390/sports7070170.
22.Webb, R., Hughes, M. G., Thomas, A. W., and
Morris, K. (2017) The ability of exercise-associated oxidative stress
to trigger redox-sensitive signalling responses, Antioxidants
(Basel), 6, doi: 10.3390/antiox6030063.
23.Merry, T. L., and Ristow, M. (2016) Mitohormesis
in exercise training, Free Radic. Biol. Med., 98,
123-130, doi: 10.1016/j.freeradbiomed.2015.11.032.
24.Ferrer, A., Caelles, C., Massot, N., and Hegardt,
F. G. (1985) Activation of rat liver cytosolic
3-hydroxy-3-methylglutaryl coenzyme A reductase kinase by adenosine
5′-monophosphate, Biochem. Biophys. Res. Commun.,
132, 497-504, doi: 10.1016/0006-291x(85)91161-1.
25.Carling, D., Clarke, P. R., Zammit, V. A., and
Hardie, D. G. (1989) Purification and characterization of the
AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase
kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities,
Eur. J. Biochem., 186, 129-136,
doi: 10.1111/j.1432-1033.1989.tb15186.x.
26.Hardie, D. G., and Carling, D. (1997) The
AMP-activated protein kinase – fuel gauge of the mammalian
cell? Eur. J. Biochem., 246, 259-273,
doi: 10.1111/j.1432-1033.1997.00259.x.
27.Hardie, D. G. (2011) Energy sensing by the
AMP-activated protein kinase and its effects on muscle metabolism,
Proc. Nutr. Soc., 70, 92-99,
doi: 10.1017/S0029665110003915.
28.Guerrieri, D., Moon, H. Y., and van Praag, H.
(2017) Exercise in a pill: the latest on exercise-mimetics, Brain
Plast., 2, 153-169, doi: 10.3233/BPL-160043.
29.Vilchinskaya, N. A., Krivoi, I. I., and Shenkman,
B. S. (2018) AMP-activated protein kinase as a key trigger for the
disuse-induced skeletal muscle remodeling, Int. J. Mol. Sci.,
19, doi: 10.3390/ijms19113558.
30.Bodur, C., Karakas, B., Timucin, A. C., Tezil,
T., and Basaga, H. (2016) AMP-activated protein kinase couples
3-bromopyruvate-induced energy depletion to apoptosis via activation of
FoxO3a and upregulation of proapoptotic Bcl-2 proteins, Mol.
Carcinogen., 55, 1584-1597, doi: 10.1002/mc.22411.
31.Shin, S., Buel, G. R., Wolgamott, L., Plas, D.
R., Asara, J. M., Blenis, J., and Yoon, S. O. (2015) ERK2 mediates
metabolic stress response to regulate cell fate, Mol. Cell,
59, 382-398, doi: 10.1016/j.molcel.2015.06.020.
32.Green, D. R., Galluzzi, L., and Kroemer, G.
(2014) Cell biology. Metabolic control of cell death, Science,
345, 1250256, doi: 10.1126/science.1250256.
33.Su, K.-H., Dai, S., Tang, Z., Xu, M., and Dai, C.
(2019) Heat shock factor 1 is a direct antagonist of AMP-activated
protein kinase, Mol. Cell, 76, 546-561,
doi: 10.1016/j.molcel.2019.08.021.
34.Sharples, A. P., Hughes, D. C., Deane, C. S.,
Saini, A., Selman, C., and Stewart, C. E. (2015) Longevity and skeletal
muscle mass: the role of IGF signalling, the sirtuins, dietary
restriction and protein intake, Aging Cell, 14, 511-523,
doi: 10.1111/acel.12342.
35.Thomson, D. M. (2018) The role of AMPK in the
regulation of skeletal muscle size, hypertrophy, and regeneration,
Int. J. Mol. Sci., 19,
doi: 10.3390/ijms19103125.
36.Ost, M., Coleman, V., Kasch, J., and Klaus, S.
(2016) Regulation of myokine expression: role of exercise and cellular
stress, Free Radic. Biol. Med., 98, 78-89,
doi: 10.1016/j.freeradbiomed.2016.02.018.
37.Eckel, J. (2019) Myokines in metabolic
homeostasis and diabetes, Diabetologia, 62, 1523-1538,
doi: 10.1007/s00125-019-4927-9.
38.Graf, C., and Ferrari, N. (2019) Metabolic health
– the role of adipo-myokines, Int. J. Mol. Sci.,
20, doi: 10.3390/ijms20246159.
39.Zhang, M., Pan, K., Liu, Q., Zhou, X., Jiang, T.,
and Li, Y. (2016) Growth differentiation factor 15 may protect the
myocardium from no-reflow by inhibiting the inflammatory-like response
that predominantly involves neutrophil infiltration, Mol. Med.
Rep., 13, 623-362, doi: 10.3892/mmr.2015.4573.
40.Zhang, Y., Moszczynski, L. A., Liu, Q., Jiang,
J., Zhao, D., Quan, D., Mele, T., McAlister, V., Jevnikar, A., Baek, S.
J., Liu, K., and Zheng, X. (2017) Over-expression of growth
differentiation factor 15 (GDF15) preventing cold ischemia reperfusion
(I/R) injury in heart transplantation through Foxo3a signaling,
Oncotarget, 8, 36531-3644,
doi: 10.18632/oncotarget.16607.
41.Lerner, L., Tao, J., Liu, Q., Nicoletti, R.,
Feng, B., Krieger, B., Mazsa, E., Siddiquee, Z., Wang, R., Huang, L.,
Shen, L., Lin, J., Vigano, A., Chiu, M. I., Weng, Z., Winston, W.,
Weiler, S., and Gyuris, J. (2016) MAP3K11/GDF15 axis is a critical
driver of cancer cachexia, J. Cachexia Sarcopenia Muscle,
7, 467-482, doi: 10.1002/jcsm.12077.
42.Jones, J. E., Cadena, S. M., Gong, C., Wang, X.,
Chen, Z., Wang, S. X., Vickers, C., Chen, H., Lach-Trifilieff, E.,
Hadcock, J. R., and Glass, D. J. (2018) Supraphysiologic administration
of GDF11 induces Cachexia in part by upregulating GDF15, Cell
Rep., 22, 1522-1530,
doi: 10.1016/j.celrep.2018.01.044.
43.Bartke, A., and Darcy, J. (2017) GH and ageing:
pitfalls and new insights, Best Pract. Res. Clin. Endocrinol.
Metab., 31, 113-125,
doi: 10.1016/j.beem.2017.02.005.
44.Rudman, D., Feller, A. G., Nagraj, H. S.,
Gergans, G. A., Lalitha, P. Y., Goldberg, A. F., Schlenker, R. A.,
Cohn, L., Rudman, I. W., and Mattson, D. E. (1990) Effects of human
growth hormone in men over 60 years old, N. Engl. J. Med.,
323, 1-6, doi: 10.1056/NEJM199007053230101.
45.Sattler, F. R. (2013) Growth hormone in the aging
male, Best Pract. Res. Clin. Endocrinol. Metab., 27,
541-555, doi: 10.1016/j.beem.2013.05.003.
46.Kim, J., and Guan, K.-L. (2019) mTOR as a central
hub of nutrient signalling and cell growth, Nat. Cell Biol.,
21, 63-71, doi: 10.1038/s41556-018-0205-1.
47.Weihrauch, M., and Handschin, C. (2018)
Pharmacological targeting of exercise adaptations in skeletal muscle:
benefits and pitfalls, Biochem. Pharmacol., 147, 211-220,
doi: 10.1016/j.bcp.2017.10.006.
48.Kaczka, P., Michalczyk, M. M., Jastrząb,
R., Gawelczyk, M., and Kubicka, K. (2019) Mechanism of action and the
effect of beta-hydroxy-beta-methylbutyrate (HMB) supplementation on
different types of physical performance – a systematic
review, J. Hum. Kinet., 68, 211-222,
doi: 10.2478/hukin-2019-0070.
49.Cruz-Jentoft, A. J. (2018)
Beta-hydroxy-beta-methyl butyrate (HMB): from experimental data to
clinical evidence in sarcopenia, Curr. Protein Pept. Sci.,
19, 668-672, doi: 10.2174/1389203718666170529105026.
50.Vyssokikh, M. Y., Holtze, S., Averina, O. A.,
Lyamzaev, K. G., Panteleeva, A. A., Marey, M. V., Zinovkin, R. A.,
Severin, F. F., Skulachev, M. V., Fasel, N., Hildebrandt, T. B., and
Skulachev, V. P. (2020) Mild depolarization of the inner mitochondrial
membrane is a crucial component of an anti-aging program, Proc.
Natl. Acad. Sci. USA, 117, 6491-6501,
doi: 10.1073/pnas.1916414117.
51.Sokolov, S. S., Markova, O. V., Nikolaeva, K. D.,
Fedorov, I. A., and Severin, F. F. (2017) Triosephosphates as
intermediates of the Crabtree effect, Biochemistry (Moscow),
82, 458-464, doi: 10.1134/S0006297917040071.
52.Chen, C., Zhou, M., Ge, Y., and Wang, X. (2020)
SIRT1 and aging related signaling pathways, Mech. Ageing Dev.,
187, 111215, doi: 10.1016/j.mad.2020.111215.
53.Santos, L., Escande, C., and Denicola, A. (2016)
Potential modulation of sirtuins by oxidative stress, Oxid. Med.
Cell. Longev., 2016, 9831825,
doi: 10.1155/2016/9831825.
54.Yamaza, H., Komatsu, T., Wakita, S., Kijogi, C.,
Park, S., Hayashi, H., Chiba, T., Mori, R., Furuyama, T., Mori, N., and
Shimokawa, I. (2010) FoxO1 is involved in the antineoplastic effect of
calorie restriction, Aging Cell, 9, 372-382,
doi: 10.1111/j.1474-9726.2010.00563.x.
55.Badreh, F., Joukar, S., Badavi, M., Rashno, M.,
and Dehesh, T. (2019) The effects of age and fasting models on blood
pressure, insulin/glucose profile, and expression of longevity proteins
in male rats, Rejuvenation Res.,
doi: 10.1089/rej.2019.2205.
56.Rodríguez-Prados, J.-C., Través, P.
G., Cuenca, J., Rico, D., Aragonés, J., Martín-Sanz, P.,
Cascante, M., and Boscá, L. (2010) Substrate fate in activated
macrophages: a comparison between innate, classic, and alternative
activation, J. Immunol., 185, 605-614,
doi: 10.4049/jimmunol.0901698.
57.Mann, G. E., Yudilevich, D. L., and Sobrevia, L.
(2003) Regulation of amino acid and glucose transporters in endothelial
and smooth muscle cells, Physiol. Rev., 83, 183-252,
doi: 10.1152/physrev.00022.2002.
58.Karavaeva, I. E., Golyshev, S. A., Smirnova, E.
A., Sokolov, S. S., Severin, F. F., and Knorre, D. A. (2017)
Mitochondrial depolarization in yeast zygotes inhibits clonal expansion
of selfish mtDNA, J. Cell Sci., 130, 1274-1284,
doi: 10.1242/jcs.197269.
59.Caldeira da Silva, C. C., Cerqueira, F. M.,
Barbosa, L. F., Medeiros, M. H. G., and Kowaltowski, A. J. (2008) Mild
mitochondrial uncoupling in mice affects energy metabolism, redox
balance and longevity, Aging Cell, 7, 552-560,
doi: 10.1111/j.1474-9726.2008.00407.x.
60.Pelletier, A., and Coderre, L. (2007) Ketone
bodies alter dinitrophenol-induced glucose uptake through AMPK
inhibition and oxidative stress generation in adult cardiomyocytes,
Am. J. Physiol. Endocrinol. Metab., 292, E1325-E1332,
doi: 10.1152/ajpendo.00186.2006.
61.Zakharova, V. V., Pletjushkina, O. Y., Galkin, I.
I., Zinovkin, R. A., Chernyak, B. V., Krysko, D. V., Bachert, C.,
Krysko, O., Skulachev, V. P., and Popova, E. N. (2017) Low
concentration of uncouplers of oxidative phosphorylation decreases the
TNF-induced endothelial permeability and lethality in mice, Biochim.
Biophys. Acta Mol. Basis Dis., 1863, 968-977,
doi: 10.1016/j.bbadis.2017.01.024.
62.Lee, Y., Heo, G., Lee, K. M., Kim, A. H., Chung,
K. W., Im, E., Chung, H. Y., and Lee, J. (2017) Neuroprotective effects
of 2,4-dinitrophenol in an acute model of Parkinson’s disease,
Brain Res., 1663, 184-193,
doi: 10.1016/j.brainres.2017.03.018.
63.Kishimoto, Y., Johnson, J., Fang, W., Halpern,
J., Marosi, K., Liu, D., Geisler, J. G., and Mattson, M. P. (2020) A
mitochondrial uncoupler prodrug protects dopaminergic neurons and
improves functional outcome in a mouse model of Parkinson’s
disease, Neurobiol. Aging, 85, 123-130,
doi: 10.1016/j.neurobiolaging.2019.09.011.
64.Coll, A. P., Chen, M., Taskar, P., Rimmington,
D., Patel, S., et al. (2020) GDF15 mediates the effects of metformin on
body weight and energy balance, Nature, 578, 444-448,
doi: 10.1038/s41586-019-1911-y.
65.Klaus, S., and Ost, M. (2020) Mitochondrial
uncoupling and longevity – a role for mitokines? Exp.
Gerontol., 130, 110796,
doi: 10.1016/j.exger.2019.110796.
66.Spiering, M. J. (2019) The mystery of metformin,
J. Biol. Chem., 294, 6689-6691,
doi: 10.1074/jbc.CL119.008628.
67.Cadenas, E., Boveris, A., Ragan, C. I., and
Stoppani, A. O. (1977) Production of superoxide radicals and hydrogen
peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c
reductase from beef-heart mitochondria, Arch. Biochem. Biophys.,
180, 248-257, doi: 10.1016/0003-9861(77)90035-2.
68.Mirphy, M. P. (2009) How mitochondria produce
reactive oxygen species, Biochem. J., 417, 1-13,
doi: 10.1042/BJ20081386.
69.Lenaz, G., Tioli, G., Falasca, A. I., and Genova,
M. L. (2016) Complex I function in mitochondrial supercomplexes,
Biochim. Biophys. Acta, 1857, 991-1000,
doi: 10.1016/j.bbabio.2016.01.013.
70.Hunt, P. R., Son, T. G., Wilson, M. A., Yu,
Q.-S., Wood, W. H., Zhang, Y., Becker, K. G., Greig, N. H., Mattson, M.
P., Camandola, S., and Wolkow, C. A. (2011) Extension of lifespan in
C. elegans by naphthoquinones that act through stress hormesis
mechanisms, PLoS One, 6, e21922,
doi: 10.1371/journal.pone.0021922.
71.Sakurai, H., and Ota, A. (2011) Regulation of
chaperone gene expression by heat shock transcription factor in
Saccharomyces cerevisiae: importance in normal cell
growth, stress resistance, and longevity, FEBS Lett.,
585, 2744-2748, doi: 10.1016/j.febslet.2011.07.041.
72.Badave, K. D., Khan, A. A., and Rane, S. Y.
(2016) Anticancer vitamin K3 analogs: a review, Anticancer Agents
Med. Chem., 16, 1017-1030,
doi: 10.2174/1871520616666160310143316.
73.Sies, H., and Jones, D. P. (2020) Reactive oxygen
species (ROS) as pleiotropic physiological signalling agents, Nat.
Rev. Mol. Cell Biol., doi: 10.1038/s41580-020-0230-3.
74.Wiel, C., Le Gal, K., Ibrahim, M. X., Jahangir,
C. A., Kashif, M., Yao, H., Ziegler, D. V., Xu, X., Ghosh, T., Mondal,
T., Kanduri, C., Lindahl, P., Sayin, V. I., and Bergo, M. O. (2019)
BACH1 stabilization by antioxidants stimulates lung cancer metastasis,
Cell, 178, 330-345,
doi: 10.1016/j.cell.2019.06.005.
75.Skulachev, M. V., and Skulachev, V. P. (2017)
Programmed aging of mammals: proof of concept and prospects of
biochemical approaches for anti-aging therapy, Biochemistry
(Moscow), 82, 1403-1422,
doi: 10.1134/S000629791712001X.
76.Isaev, N. K., Stelmashook, E. V., Genrikhs, E.
E., Korshunova, G. A., Sumbatyan, N. V., Kapkaeva, M. R., and
Skulachev, V. P. (2016) Neuroprotective properties of
mitochondria-targeted antioxidants of the SkQ-type, Rev.
Neurosci., 27, 849-855,
doi: 10.1515/revneuro-2016-0036.
77.Baksheeva, V. E., Gancharova, O. S., Tiulina, V.
V., Iomdina, E. N., Zamyatnin, A. A. Jr., Philippov, P. P., Zernii, E.
Y., and Senin, I. I. (2018) Iatrogenic damage of eye tissues: current
problems and possible solutions, Biochemistry (Moscow),
83, 1563-1574, doi: 10.1134/S0006297918120143.
78.Sies, H., Berndt, C., and Jones, D. P. (2017)
Oxidative stress, Annu. Rev. Biochem., 86, 715-748,
doi: 10.1146/annurev-biochem-061516-045037.
79.Marinho, H. S., Real, C., Cyrne, L., Soares, H.,
and Antunes, F. (2014) Hydrogen peroxide sensing, signaling and
regulation of transcription factors, Redox Biol., 2,
535-562, doi: 10.1016/j. redox.2014.02.006.
80.Borisova-Mubarakshina, M. M., Vetoshkina, D. V.,
and Ivanov, B. N. (2019) Antioxidant and signaling functions of the
plastoquinone pool in higher plants, Physiol. Plant.,
166, 181-198, doi: 10.1111/ppl.12936.
81.Borisova-Mubarakshina, M. M., Naydov, I. A., and
Ivanov, B. N. (2018) Oxidation of the plastoquinone pool in chloroplast
thylakoid membranes by superoxide anion radicals, FEBS Lett.,
592, 3221-3228, doi: 10.1002/1873-3468.13237.
82.Severin, F. F., and Skulachev, V. P. (2009)
Programmed cell death as a target to interrupt the aging program,
Adv. Gerontol., 22, 37-48.
83.NCD Countdown 2030 collaborators (2018) NCD
Countdown 2030: worldwide trends in non-communicable disease mortality
and progress towards Sustainable Development Goal target 3.4,
Lancet, 392, 1072-1088,
doi: 10.1016/S0140-6736(18)31992-5.
84.Gong, J. B., Yu, X. W., Yi, X. R., Wang, C. H.,
and Tuo, X. P. (2018) Epidemiology of chronic noncommunicable diseases
and evaluation of life quality in elderly, Aging Med., 1,
64-66, doi: 10.1002/agm2.12009.