Involvement of SASH1 in the Maintenance of Stable Cell–Cell Adhesion
A. S. Ilnitskaya1, I. Y. Zhitnyak1, and N. A. Gloushankova1,a*
1Blokhin National Medical Research Center of Oncology, Ministry of Health of the Russian Federation, 115478 Moscow, Russia* To whom correspondence should be addressed.
Received April 1, 2020; Revised April 29, 2020; Accepted May 3, 2020
SASH1 is an adapter and signaling protein that contains SH3 and SAM domains responsible for protein–protein interactions. SASH1 downregulation has been observed in many tumors. We examined localization of SASH1 in cultures of normal IAR-20 epithelial cells and HT-29 colorectal cancer cells using immunofluorescence staining and confocal microscopy. IAR-20 normal epithelial cells and HT-29 cells with epithelial phenotype formed stable linear adherens junctions (AJs) associated with circumferential actin bundles. In both IAR-20 and HT-29 cells, SASH1 was co-localized with zones of circumferential actin bundles and linear AJs. SASH1 was not detected in lamellipodia. IAR-20 and HT-29 cells treated with Epidermal Growth Factor underwent epithelial-mesenchymal transition (EMT). We observed significant differences in the course of EMT between IAR-20 and HT-29 cultures. IAR-20 cells underwent partial EMT acquiring migratory phenotype but retaining E-cadherin in unstable radial AJs. SASH1 was present in these contacts. Disappearance of AJs was observed in HT-29 cell undergoing a complete EMT, which also resulted in disruption of stable cell–cell adhesion. SASH1 was lost from the zones of cell–cell interaction. SASH1 depletion by means of RNA interference in IAR-20 cells led to destruction of stable linear AJs and acquisition of mesenchymal phenotype by some of the cells. These data indicate involvement of SASH1 in the maintenance of stable cell–cell adhesion.
KEY WORDS: SASH1, cell–cell adhesion, E-cadherin, epithelial-mesenchymal transitionDOI: 10.1134/S0006297920060036
Abbreviations: AJs, adherens junctions; EGF, epidermal growth factor; EMT, epithelial-mesenchymal transition.
INTRODUCTION
SASH1 gene was originally described as a gene whose expression was downregulated in breast cancer [1]. SASH1 is expressed in multiple tissues, with the exception of dendritic cells and lymphocytes, and SASH1 has been suggested as a candidate tumor suppressor gene. Downregulation of SASH1 expression was found in 74% of breast cancer, in lung carcinoma, thyroid carcinoma, and hepatocarcinoma [1, 2]. SASH1 is considered a prognostic marker in breast, colon, ovarian, and cervical carcinoma, strong downregulation of SASH1 expression was found at advanced stages of cancer. High level of SASH1 expression, however, was maintained in adenoma and at earlier stages of carcinoma [1, 3-5].
SASH1 protein contains the central region with nuclear localization signal (NLS), two sterile alpha motif (SAM) domains, and Src homology domain 3 (SH3) that mediate protein–protein interactions [1]. The presence of SH3 domain indicates that SASH1 could have scaffold and adapter functions, participate in signaling, and interact with membrane proteins. Overexpression of SASH1 inhibited migratory and invasive activity of hepatocarcinoma and gastric carcinoma cells, strengthened cell-extracellular matrix adhesion, suppressed the levels of mesenchymal markers, N-cadherin and vimentin, and increased the level of E-cadherin [6, 7]. Knockdown of SASH1 in HCT116 cells using the CRISPR-Cas9 editing system induced down-regulation of E-cadherin, up-regulation of vimentin and epithelial-mesenchymal transition (EMT)-promoting transcription factor Zeb1. Loss of SASH1 resulted in activation of migratory and invasive activity of cells in vitro and enhanced metastatic outgrowth of cells in vivo in orthotopic xenograft mouse model [8]. It was shown that SASH1 interacts with N-terminal SH3 domain of CRKL. It has been suggested that CRKL induces activation of SRC/FAK signaling, and thus EMT [9]. CRK proteins can also recruit guanine nucleotide exchange factors into close proximity to the membrane, inducing activation of the small GTPases Rap1 and Rac1 [10]. It is well known that the small GTPase Rac1 plays a key role in cell migration. Active Rac activates the ARP2/3 complex by WAVE that nucleates polymerization of actin filament network in lamellipodia at the leading edge of a migrating cell [11]. Although numerous data confirmed the effect of changes in the content of SASH1 on functional characteristics of the cells, intracellular localization of SASH1 have not been fully examined yet. Martini et al. [12] reported that SASH1 accumulated in lamellipodia and was colocalized with cortactin that interacted with Arp2/3 complex and stabilized the structure of actin network thus facilitating effective cell migration [13].
Acquisition of migratory activity by epithelial cells is a key characteristic of EMT – the program that plays a leading role in the development and wound healing. Cancer cells use the EMT program to initiate the invasion-metastasis cascade. During EMT, epithelial cells lose apical-basal polarity and stable cell–cell adhesion thus acquiring migratory phenotype [14, 15]. It is presumed that EMT-inducing transcription factors, such as Snail, Twist or Zeb trigger EMT and associated suppression of E-cadherin expression, which results in weakening of cell–cell adhesion. We recently showed that loss of stable cell–cell adhesion could be also be due to reorganization of the actin cytoskeleton and replacement of linear E-cadherin-based adherens junctions (AJs) with dynamic and unstable radial AJs [16]. It was demonstrated that circumferential β-actin bundles [17] were disrupted at the early stages of EMT and this led to reorganization of AJs and weakening of cell–cell adhesion. In the present study, we examined localization of SASH1 in the zones of cell–cell interaction of epithelial cells. We also studied redistribution of SASH1 during destruction of stable AJs and circumferential actin bundle in the course of EMT.
MATERIALS AND METHODS
Cell culture. Normal immortalized epithelial cell line IAR-20 was isolated from rat liver at the International Cancer Agency by Montesano and colleagues [18]. Human colorectal adenocarcinoma line HT-29 (ATCC, USA) was also used in the work. Cells (1 × 105) were seeded into Petri dishes containing glass coverslips or into glass-bottom Petri dishes (MatTek Corporation, USA) and incubated in Dulbecco’s modified Eagle medium (DMEM) (Sigma, USA) with 10% fetal calf serum (PAA Laboratories, Austria) for 24 h. Next the medium was replaced with fresh DMEM containing 1% fetal calf serum followed by incubation for 20 h. To induce EMT, stock solution of epidermal growth factor (EGF, Sigma) was added to IAR-20 and HT-29 cells to concentration 40 and 50 ng/ml, respectively.
Immunofluorescence microscopy and DIC videomicroscopy. Following antibodies were used for immunofluorescence staining: mouse monoclonal anti-E-cadherin (clone 36, 1 : 200; BD Transduction Labs, USA), rabbit anti-SASH1 (clone 266A, 1 : 100; Bethyl Laboratories, USA), mouse monoclonal anti-β-actin (clone 4C2, 1 : 100; Merck, Millipore, USA), mouse monoclonal anti-cortactin (clone 4F11, 1 : 200; Sigma), goat anti-mouse IgG1, IgG2a, and goat anti-mouse IgG and anti-rabbit IgG conjugated with Alexa Fluor488, Alexa Fluor594 or Alexa Fluor647 (1 : 200; Jackson ImmunoResearch, USA). Cells on coverslips were fixed for 15 min with a mixture of methanol/acetone (1/1) at –10°C and incubated with primary antibodies. After washing with phosphate buffer (1× PBS, pH 7.4), fixed specimens were incubated with secondary antibodies. The samples were examined with a Leica TCS SP5 confocal microscope using an HDX PL APO 63×/1.3 lens (Leica Microsystems, Switzerland) and an Axioplan Zeiss epifluorescence microscope using a Plan-Neofluar 100×/1.3 lens (Carl Zeiss, Germany). For DIC videomicroscopy, the culture medium was replaced by DMEM/F12 with L-glutamine, HEPES with 1% fetal calf serum, but without Phenol Red 20 min before imaging. One hour after beginning of the observation, EGF was added to the culture medium to induce EMT. Live-cell imaging was performed using a Nikon Eclipse-Ti microscope with a PlanFluor40× objective lens, ORCA-ER digital camera (Hamamatsu Photonics, Japan), and NIS-Elements AR 3.22 software (Nikon, Japan). Images were captured at a rate 1 frame/minute for 6 h (for IAR-20 cell culture) and 1 frame/10 min for 10 h (for HT-29 cell culture).
RNA interference. ON-TARGETplus SMARTpool rat Sash1 siRNA (50 nM) and DharmaFECT1 (Dharmacon, USA) as a transfection agent were used to suppress SASH1. GFP siRNA was used as a negative control. Cultures were incubated in DMEM with 10% serum for 48 h followed by cell lysis and examination by immunoblotting. Coverslips with transfected cells were also fixed for immunofluorescence staining.
Immunoblotting. Cells were lysed with RIPA lysis buffer (50 mM Tris-HCl, pH 7.4 (MP Biomedicals, France); 150 mM NaCl (Sigma); 2 mM EDTA (Sigma); 1% NP-40 (Fluka, USA); 0.1% SDS (AppliChem, Spain) supplemented with 0.25 mM Na3VO4, 1 mM DTT, 10 mM NaF and a protease inhibitor cocktail (Sigma, USA). Samples were mixed with 5× application buffer (250 mM Tris-HCl, pH 6.8; 10% SDS, 30% (v/v) glycerol, 5% β-mercaptoethanol, 0.02% bromophenol blue) and incubated at 95°C for 10 min. Next, samples were loaded onto a 10% SDS-polyacrylamide gel according to SDS-PAGE protocol (Bio-Rad, USA). After electrophoresis resolved proteins were transferred to Amersham Hybond-P PVDF membranes (GE Healthcare). Membranes were blocked with a 5% milk solution (Fluka) in phosphate buffer (1× PBS pH 7.4) with 0.1% (v/v) Tween-20 (AppliChem, Spain) for 1 h followed by incubation with primary antibodies for 16 h at 4°C. After washing with phosphate buffer with 0.1% (v/v) Tween-20, membranes were incubated with secondary peroxidase conjugated antibodies for 1 h at room temperature. Monoclonal anti-actin antibodies (clone C4, 1 : 1000; Merck) were used as a loading control. Protein bands were detected using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, USA) and images were captured with an ImageQuant LAS 4000 (GE Healthcare). For densitometric analysis of blots ImageJ [imagej.net] software was used. Optical densities of SASH1 bands were normalized to a marker protein, actin. Student’s t-test was used for statistical analysis of the results of three experiments, and the data were presented as mean values ± SEM.
RESULTS
Distribution of SASH1 in IAR-20 normal epithelial cells and in HT-29 colorectal cancer cells was analyzed with confocal microscopy. The cells of these lines maintained an epithelial phenotype and formed islands in a sparse culture, and monolayers in a dense culture. Triple immunofluorescence staining of E-cadherin, actin, and SASH1 was performed (Fig. 1a). Confocal microscopy showed that IAR-20 cells formed linear AJs associated with circumferential actin bundles, and that SASH1 accumulated in AJs. SASH1 was also detected in the zones of the marginal actin bundles. SASH1 antibodies provided good staining of the samples fixed with methanol/acetone mixture, however, this also resulted in non-specific fluorescence of secondary antibodies (Fig. 1b). Distinct fluorescence of cytoplasm in the far-red channel of the confocal microscope was not associated with SASH1 and was due to the sorption of Alexa647-labeled secondary anti-rabbit IgG antibodies at intracellular organelles. Nonspecific fluorescence of secondary anti-mouse IgG isotype antibodies used for simultaneous staining of E-cadherin and β-actin was also detected in the cytoplasm. Nonspecific staining was not significantly pronounced at the periphery of the cells, which allowed us to examine cell–cell-boundaries and cell edges in detail.
Fig. 1. SASH1 in IAR-20 epithelial cells. a) Confocal microscopy: E-cadherin (red channel), β-actin (green channel), SASH1 (far red channel). Arrowhead indicates cell–cell contact enlarged in the boxed region. SASH1 accumulates in the zones of AJs. b) Confocal microscopy. Control of secondary antibodies. Nonspecific fluorescence of secondary antibodies. Red channel is anti-mouse IgG2a labeled with Alexa594, green channel is anti-mouse IgG1 labeled with Alexa488, far red channel is anti-rabbit IgG labeled with Alexa647. c) Fluorescence staining for cortactin, E-cadherin and SASH1. SASH1 is detected in AJs and in the zone of the marginal actin bundle at the base of lamella. Lamellipodia in which SASH1 is absent (indicated with arrowhead), enlarged in the boxed region. Arrow indicates accumulation of SASH1 in the zone of marginal actin bundle enlarged in the boxed region. Scale bar: 10 µm.
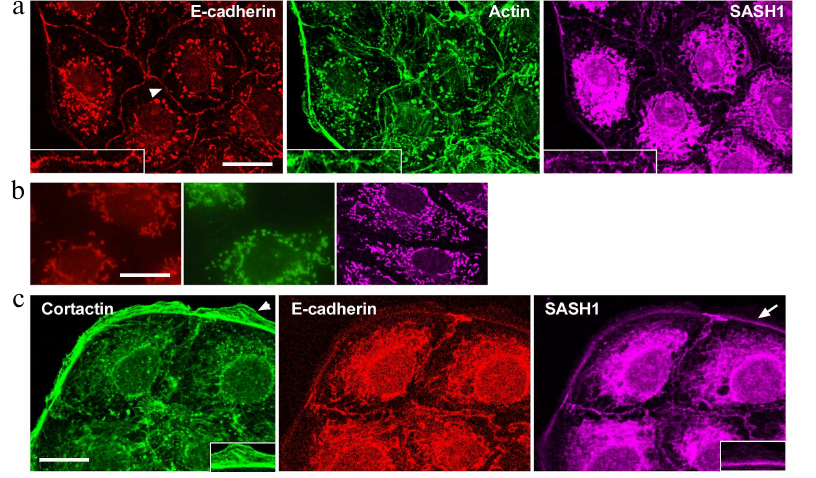
Martini et al. [12] stated that SASH1 was detected in lamellipodia and colocalized with cortactin – a protein linked to actin network. We showed, that there is no colocalization of cortactin and SASH1 in both IAR-20 and HT-29 cells. In IAR-20 cells, cortactin was present in actin network of lamellipodia at the leading cell edge and colocalized with a marginal actin bundle (Fig. 1c), whereas SASH1 was absent from the zones of lamellipodia. Distribution of SASH1 at the leading cell edge was limited to the marginal actin bundle.
We demonstrated previously that IAR-20 cells treated with EGF undergo EMT disrupting stable cell–cell contacts and acquiring the ability to migrate [16] (Fig. 2a).
Fig. 2. IAR-20 epithelial cells in the presence of EGF. a) DIC video microscopy. Before the addition of EGF, IAR-20 cells form islands. After the addition of EGF (0 min), the cells undergo EMT: protrusive activity is induced, cell–cell contacts are disrupted, cell migration is activated. Migrating cells can form contacts with neighboring cells and these contacts are unstable. Arrowheads: zones of cell–cell contact disruption, arrows: newly formed contacts. b, c) Fluorescence staining of β-actin, E-cadherin, SASH1 in IAR-20 cells. b) Ten-minutes incubation with EGF. The appearance of lamellipodia at the cell–cell-boundaries. SASH1 is not detected in lamellipodia. c) Thirty-minutes incubation with EGF. SASH1 is present in radial E-cadherin-based AJs. The cell–cell boundaries indicated with arrowheads are enlarged in boxed regions. Scale bar: 10 µm.
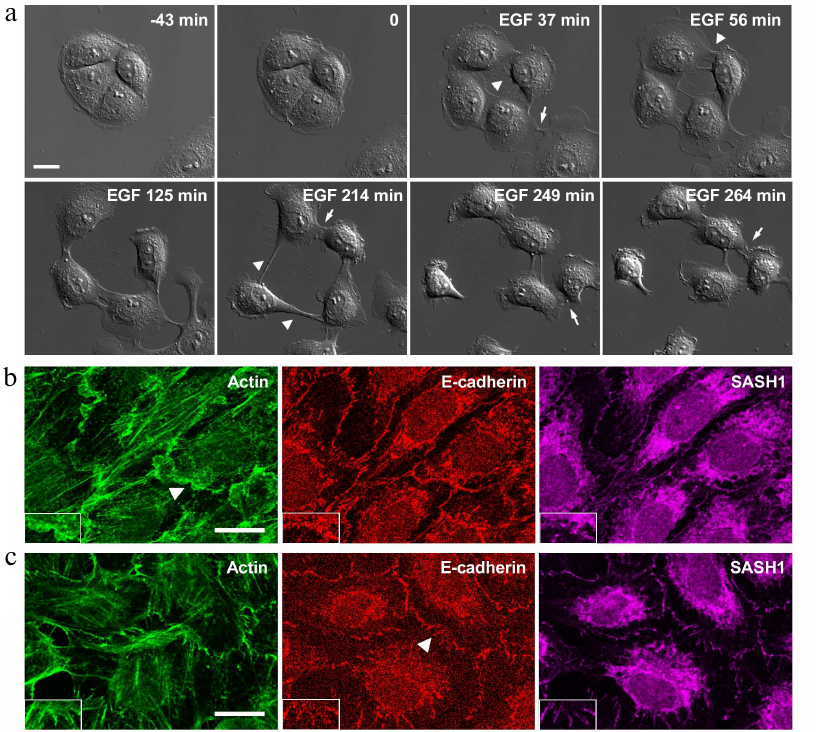
In the presence of EGF, IAR-20 cells can migrate both individually and collectively and cells can establish contacts with neighboring cells; such contacts, however, are unstable. Studying the early stages of EMT, we established that weakening of cell–cell adhesion in IAR-20 cells was not associated with downregulation of the E-cadherin expression or its reduced accumulation at the cell–cell boundaries. We showed that weakening of cell–cell adhesion coincided with disruption of the circumferential actin bundle associated with linear AJs, the appearance of lamellipodia at the cell–cell-boundaries, and replacement of linear AJs with unstable radial AJs, which were also formed by E-cadherin. In this connection we decided to explore changes in the distribution of SASH1 in IAR-20 cells undergoing EMT. It was found out that in parallel with AJ reorganization, localization of SASH1 in the zones of cell–cell interaction also changed. Confocal microscopy showed that SASH1 was absent from lamellipodia both on the leading cell edge and at the cell–cell boundaries. At the same time SASH1 was detected in the newly formed radial AJs (Fig. 2, b and c).
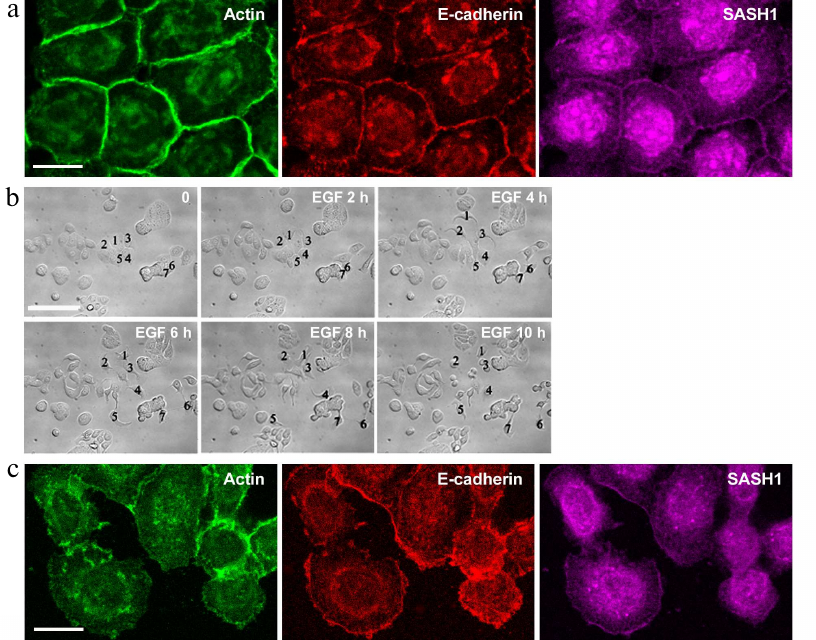
We also studied distribution of SASH1 in the cells of HT-29 colorectal adenocarcinoma line. HT-29 cells maintain an epithelial phenotype and form stable E-cadherin-based AJs (Fig. 3a). Such linear AJs were associated with the circumferential actin bundle. SASH1 in HT-29 cells accumulated in the zones of linear AJs. HT-29 cells treated with EGF underwent EMT. Unlike in the case of IAR-20 epitheliocytes, HT-29 cells completed the EMT process during which the cells lost cell–cell contacts and acquired ability to migrate individually (Fig. 3b). Immunofluorescence staining showed complete disruption of AJs in HT-29 cells in the presence of EGF. E-cadherin left the contacts and accumulated in endosomes. SASH1 also left the zones of cell–cell interaction in parallel with disruption of AJs. In some cells with actin filaments accumulated at the leading cell edge SASH1 was present as a thin line in this zone (Fig. 3c).
Fig. 3. SASH1 in HT-29 carcinoma cells. a) Fluorescence staining of control culture for E-cadherin, β-actin, and SASH1. Confocal microscopy. Cells form linear AJs associated with the circumferential actin bundles. SASH1 accumulates in the zones of AJs. Scale bar: 10 µm. b, c) EGF-induced EMT. b) DIC videomicroscopy. Before the addition of EGF, HT-29 cells form islands. EGF-treated cells undergo EMT: lose stable cell–cell contacts and acquire migratory activity. Numbers indicate location of the cells. Scale bar: 50 µm. c) Eight-hour incubation with EGF. Fluorescence staining for E-cadherin, β-actin, and SASH1. Complete disruption of AJs and endocytosis of E-cadherin. SASH1 is absent at the cell–cell boundaries but is present at the cell edges. Scale bar: 10 µm.
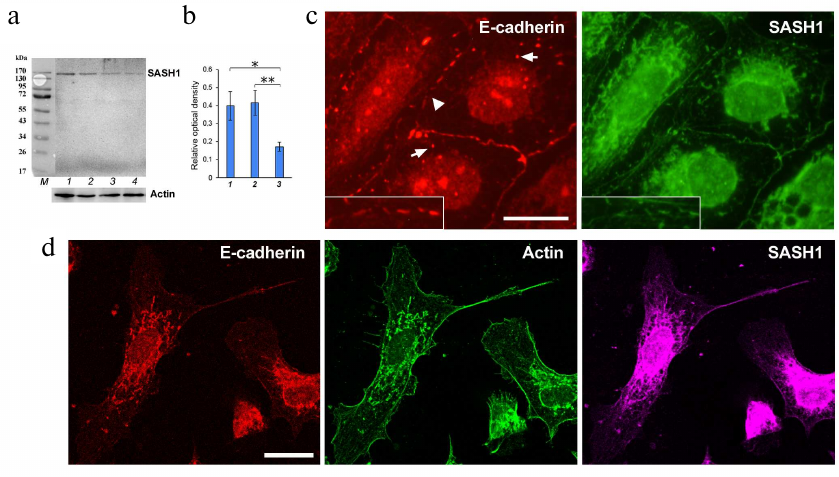
Thus, we observed close association between SASH1 and AJs implying that SASH1 might participate in the maintenance of AJs. We decided to find out how SASH1 suppression affected cell morphology and cell–cell adhesion. ON-TARGETplus SMARTpool rat SASH1 siRNAs were used for SASH1 suppression (Fig. 4, a and b). Analysis of immunofluorescence revealed that suppression of SASH1 in IAR-20 cells resulted in significant alterations in the structure of cell–cell contacts. Stable linear E-cadherin AJs characteristic for IAR-20 epithelial cells were destroyed 48 h after transfection with SASH1 siRNA. Single punctate or short radial AJs were observed at the cell–cell boundaries (Fig. 4c). Endosomes containing E-cadherin were detected in cytoplasm. Many cells in the culture lost their epithelial phenotype and acquired a mesenchymal phenotype; and cell islands often underwent scattering (Fig. 4d). Hence, it was shown that decrease of the SASH1 content in cells reduced the stability of cell–cell adhesion, which affected cell morphology and led to acquisition of a migratory phenotype by the cells.
Fig. 4. Suppression of SASH1 in IAR-20 cells. a) Western blot analysis of SASH1 in culture (48 h after transfection of SASH1 siRNA). Staining of the full-sized membrane for SASH1 and for total actin. Lines: 1) GFP siRNA (negative control); 2) control culture; 3, 4) SASH1 siRNA. b) Densitometric analysis of blots from three experiments. * p < 0.05, ** p < 0.01. Bars: 1) GFP siRNA (negative control); 2) control culture; 3) SASH1 siRNA. c, d) Fluorescence staining of cells (48 h after transfection of SASH1 siRNA). c) Epifluorescence microscopy of E-cadherin and SASH1. At the cell–cell boundaries, E-cadherin accumulates in the sparse punctate AJs (arrowhead). Endosomes contain E-cadherin (arrows). d) Confocal microscopy of E-cadherin, β-actin, SASH1. Cells acquired a mesenchymal phenotype. Scale bar: 10 µm.
DISCUSSION
We demonstrated for the first time that SASH1 was involved in the maintenance of stable cell–cell adhesion. Our data revealed that suppression of SASH1 induced by RNA-interference resulted in disruption of the stable E-cadherin-based AJs and shifted epithelial cells toward a mesenchymal phenotype. These data are in agreement with the data of Franke et al. [8] who described EMT in hepatocarcinoma cells, where deficiency of SASH1 was induced by CRISPR-Cas9. Interaction of SASH1 with activated Rac was demonstrated in earlier studies using the yeast two hybrid screening [19]. Based on these results, SASH1 could be considered as the Rac effector. Nevertheless, the available data on involvement of Rac in AJ formation are contradictory. On the one hand, it is considered that active Rac stabilizes AJs recruiting actin to E-cadherin adhesive homodimers [20]. On the other hand, high Rac activity is detected in the zones of cell–cell interaction only at the early stages of formation of stable AJs during lamellipodia interactions and it diminishes significantly during lateral expansion of linear AJs [21]. Recently we discovered that in IAR-20 cell at the early stages of EMT disruption of circumferential actin bundle and replacement of stable AJs by unstable radial AJs is accompanied by Arp2/3-dependent polymerization of actin network and formation of lamellipodia at the cell–cell boundaries, i.e., activation of Rac. These data suggest that Rac is not involved in the maintenance of AJ stability. Growth factor induced activation of Rac, on the contrary, leads to formation of lamellipodia at the cell–cell boundaries, disappearance of contact paralysis, and weakening of cell–cell adhesion [16]. Based on our data, we can hypothesize that SASH1 could not be a Rac-GTP effector, but its negative regulator stabilizing cell–cell adhesion. Earlier it was shown that SASH1 interacted with N-terminal SH3 domain of CRKL and activated oncogenic SRC/FAK signaling [9]. Moreover, overexpression of CRK (CRK II and CRKL) proteins resulted in Rac activation, formation of lamellipodia at the leading cell edge, disruption of AJs, and scattering of epithelial islands [22]. It is likely that in the zones of cell–cell boundaries, SASH1, as a scaffold protein, recruits CRKL, Rac and Rac GAP, and their interaction results in suppression of Rac activity and stabilization of cell–cell contacts. Depletion of SASH1 stimulates oncogenic activity of CRKL and this leads to activation of Rac and disruption of cell–cell adhesion. Further studies are required to confirm this hypothesis.
Considering that SASH1 has nuclear localization signal sequence (NLS) and localizes at the cell–cell boundaries, SASH1 could be associated with contact inhibition of proliferation typical for normal epithelial cells. Such regulatory function was described for the multifunctional protein Merlin that was not only associated with circumferential actin bundle and AJs, but also negatively regulated EGFR and Rac/PAK signaling and facilitated export of the transcription coactivators YAP/TAZ from the nucleus in high-density monolayers of epithelial cells [23]. SASH1 can also be a key protein whose ability to form complexes with proteins both on the cell membrane and in the nucleus ensures its significant role in tumor suppression by combining its negative effects on cells’ migratory and proliferative activity.
Funding. The work was financially supported by the Russian Foundation for Basic Research (project No. 18-54-16005). The study of EMT was funded by the Russian Science Foundation (project No. 16-15-10288).
Conflict of interest. The authors declare no conflict of interest in financial or any other sphere.
Compliance with ethical norms. This article does not contain any studies with human participants or animals performed by any of the authors.
REFERENCES
1.Zeller, C., Hinzmann, B., Seitz, S., Prokoph, H.,
Burkhard-Goettges, E., Fischer, J., Jandrig, B., Schwarz, L.-E.,
Rosenthal, A., and Scherneck, S. (2003) SASH1: a candidate tumor
suppressor gene on chromosome 6q24.3 is downregulated in breast cancer,
Oncogene, 22, 2972-2983,
doi: 10.1038/sj.onc.1206474.
2.Peng, L., Wei, H., and Liren, L. (2014) Promoter
methylation assay of SASH1 gene in hepatocellular carcinoma, J.
BUON, 19, 1041-1047.
3.Rimkus, C., Martini, M., Friederichs, J.,
Rosenberg, R., Doll, D., Siewert, J. R., Holzmann, B., and Janssen, K.
P. (2006) Prognostic significance of downregulated expression of the
candidate tumour suppressor gene SASH1 in colon cancer, Br. J.
Cancer, 95, 1419-1423, doi: 10.1038/sj.bjc.6603452.
4.Ren, X., Liu, Y., Tao, Y., Zhu, G., Pei, M., Zhang,
J., and Liu, J. (2016) Downregulation of SASH1 correlates with tumor
progression and poor prognosis in ovarian carcinoma, Oncol.
Lett., 11, 3123-3130, doi: 10.3892/ol.2016.4345.
5.Xie, J., Zhang, W., Zhang, J., and Luan, Y.-F.
(2017) Downregulation of SASH1 correlates with poor prognosis in
cervical cancer, Eur. Rev. Med. Pharm. Sci., 21,
3781-3786.
6.He, P., Zhang, H., Sun, C., Chen, C., and Jiang, H.
(2016) Overexpression of SASH1 inhibits the proliferation, invasion,
and EMT in hepatocarcinoma cells, Oncol. Res., 24, 25-32,
doi: 10.3727/096504016X14575597858609.
7.Zong, W., Yu, C., Wang, P., and Dong, L. (2016)
Overexpression of SASH1 inhibits TGF-b1-induced EMT in gastric cancer
cells, Oncol. Res., 24, 17-23,
doi: 10.3727/096504016X14570992647203.
8.Franke, F. C., Müller, J., Abal, M., Medina,
E. D., Nitsche, U., Weidmann, H., Chardonnet, S., Ninio, E., and
Janssen, K. P. (2018) The tumor suppressor SASH1 interacts with the
signal adaptor CRKL to inhibit epithelial-mesenchymal transition and
metastasis in colorectal cancer, Cell. Mol. Gastroenterol.
Hepatol., 7, 33-53,
doi: 10.1016/j.jcmgh.2018.08.007.
9.Franke, F. C., Slusarenko, B. O., Engleitner, T.,
Johannes, W., Laschinger, M., Rad, R., Nitsche, U., and Janssen, K. P.
(2020) Novel role for CRK adaptor proteins as essential components of
SRC/FAK signaling for epithelial-mesenchymal transition and colorectal
cancer aggressiveness, Int. J. Cancer, 146, doi:
10.1002/ijc.32955.
10.Feller, S. (2001) Crk family adaptors-signalling
complex formation and biological roles, Oncogene, 20,
6348-6371, doi: 10.1038/sj.onc.1204779.
11.Krause, M., and Gautreau, A. (2014) Steering cell
migration: lamellipodium dynamics and the regulation of directional
persistence, Nat. Rev. Mol. Cell Biol., 15, 577-590,
doi: 10.1038/nrm3861.
12.Martini, M., Gnann, A., Scheikl, D., Holzmann,
B., and Janssen, K. P. (2011) The candidate tumor suppressor SASH1
interacts with the actin cytoskeleton and stimulates cell–matrix
adhesion, Int. J. Biochem. Cell. Biol., 43, 1630-1640,
doi: 10.1016/j.biocel.2011.07.012.
13.Rottner, K., and Stradal, T. (2016) How distinct
Arp2/3 complex variants regulate actin filament assembly, Nat. Cell
Biol., 18, 1-3, doi: 10.1038/ncb3293.
14.Lamouille, S., Xu, J., and Derynck, R. (2014)
Molecular mechanisms of epithelial-mesenchymal transition, Nat. Rev.
Mol. Cell Biol., 15, 178-196, doi: 10.1038/nrm3758.
15.Nieto, M. A., Huang, R. Y., and Jackson, R. A.,
and Thiery, J. P. (2016) EMT: 2016, Cell, 166, 21-45,
doi: 10.1016/j.cell.2016.06.028.
16.Zhitnyak, I. Y., Rubtsova, S. N., Litovka, N. I.,
and Gloushankova, N. A. (2020) Early events in actin cytoskeleton
dynamics and E-cadherin-mediated cell–cell adhesion during
epithelial-mesenchymal transition, Cells, 9, pii: E578,
doi: 10.3390/cells9030578.
17.Dugina, V., Zwaenepoel, I., Gabbiani, G.,
Clément, S., and Chaponnier, C. (2009) β- and
γ-Cytoplasmic actins display distinct distribution and functional
diversity, J. Cell Sci., 122, 2980-2988,
doi: 10.1242/jcs.041970.
18.Montesano, R., Saint Vincent, L., Drevon, C., and
Tomatis, L. (1975) Production of epithelial and mesenchymal tumors with
rat liver cells transformed in vitro, Int. J. Cancer,
16, 550-558, doi: 10.1002/ijc.2910160405.
19.Lomakina, M. E., Polesskaya, A., Aleksandrova, A.
Yu., and Gotro, A. (2016) Search for new cell viral regulators among
recently discovered Rac GTPase binding partners, Adv. Mol.
Oncol., 3, 20.
20.McCormack, J., Welsh, N. J., and Braga, V. M.
(2013) Cycling around cell–cell adhesion with Rho GTPase
regulators, J. Cell Sci., 126, 379-391,
doi: 10.1242/jcs.097923.
21.Yamada, S., and Nelson, W. J. (2007) Localized
zones of Rho and Rac activities drive initiation and expansion of
epithelial cell–cell adhesion, J. Cell Biol., 178,
517-527, doi: 10.1083/jcb.200701058.
22.Lamorte, L., Royal, I., Naujokas, M., and Park,
M. (2002) Crk adapter proteins promote an epithelial-mesenchymal-like
transition and are required for HGF-mediated cell spreading and
breakdown of epithelial adherens junctions, Mol. Biol. Cell,
13, 1449-1461, doi: 10.1091/mbc.01-10-0477.
23.Furukawa, K. T., Yamashita, K., Sakurai, N., and
Ohno, S. (2017) The epithelial circumferential actin belt regulates
YAP/TAZ through nucleocytoplasmic shuttling of merlin, Cell
Rep., 20, 1435-1447, doi: 10.1016/j.
celrep.2017.07.032.