Highly-Active Recombinant Formate Dehydrogenase from Pathogenic Bacterium Staphylococcus aureus: Preparation and Crystallization
A. A. Pometun1,2,3#, K. M. Boyko2#, T. S. Yurchenko1,2, A. Yu. Nikolaeva2,4, I. S. Kargov2,3, D. L. Atroshenko1,2,3, S. S. Savin1,2,3, V. O. Popov2,4, and V. I. Tishkov1,2,3,a*
1Lomonosov Moscow State University, Faculty of Chemistry, 119991 Moscow, Russia2Bach Institute of Biochemistry, Federal Research Centre “Fundamentals of Biotechnology” of the Russian Academy of Sciences, 119071 Moscow, Russia
3Innovations and High Technologies MSU Ltd., 109559 Moscow, Russia
4Kurchatov Institute National Research Center, 123182 Moscow, Russia
# These authors contributed equally to the work.
* To whom correspondence should be addressed.
Received April 16, 2020; Revised May 2, 2020; Accepted May 2, 2020
NAD+-dependent formate dehydrogenase from Staphylococcus aureus (SauFDH) is one of the key enzymes responsible for the survival of this pathogen in the form of biofilms. 3D structure of the enzyme might be helpful in the search for highly specific SauFDH inhibitors that can be used as antibacterial agents exactly against S. aureus biofilms. Here, we prepared a recombinant SauFDH in Escherichia coli cells with a yield of 1 g target protein per liter medium. The developed procedure for the enzyme purification allowed to obtain 400 mg of homogenous enzyme with 61% yield. The specific activity of the purified recombinant SauFDH was 20 U per mg protein, which was 2 times higher than the previously reported activities of formate dehydrogenases. We also found crystallization conditions in the course of two rounds of optimization and obtained 200- and 40-µm crystals for the SauFDH apo- and holoenzymes, respectively. X-ray analysis using synchrotron X-ray sources produced diffraction data sufficient for solving the three-dimensional structures of the apo- and holoenzymes with the resolution of 2.2 and 2.7 Å, respectively. Crystals of the apo- and holoforms of SauFDH had different crystal space groups, which suggest coenzyme binding in the SauFDH holoenzyme.
KEY WORDS: formate dehydrogenase, Staphylococcus aureus, expression, purification, crystallization, X-ray diffraction analysisDOI: 10.1134/S0006297920060061
Abbreviations: FDH formate dehydrogenase; SauFDH, formate dehydrogenase from Staphylococcus aureus; PseFDH, formate dehydrogenase from Pseudomonas sp. 101
INTRODUCTION
NAD+-dependent formate dehydrogenase (FDH) has been found in bacteria, yeast, fungi, and plants. The major function of this enzyme in pathogenic microorganisms and plants is supplying the energy to the cells in a form of reduced coenzyme NADH under stress conditions. Advances in high-throughput sequencing have significantly increased the number of genome sequences of various pathogens in the databases. FDH genes have been annotated in the genomes of almost all pathogenic bacteria and yeast [1-3], including FDH from Staphylococcus aureus (SauFDH). S. aureus is one of the most dangerous human pathogens that causes a wide variety of diseases, such as pneumonia, toxic shock syndrome, sepsis, and others, and often identified as a cause of nosocomial (hospital) infections. Staphylococci are most dangerous when growing in biofilms form, as they display high resistance to common antibacterial agents in this state. Transcriptome analysis [4] demonstrated that the level of SauFDH expression in S. aureus biofilms was 20-fold higher than in planktonic cells; moreover, the total amount of SauFDH mRNA was the third largest among RNAs for other proteins [4], suggesting that SauFDH is a key enzyme ensuring survival of S. aureus in the biofilms. Therefore, SauFDH might be a promising target in the fight against staphylococcal (staph) biofilms. However, development of highly specific SauFDH inhibitors requires availability of the enzyme 3D structure (structure-based drug design).
SauFDH is also of considerable interest for the fundamental enzymology. NAD+-dependent FDH is a very conserved enzyme that consists of two identical polypeptide chains without additional groups in its molecule. The homology between FDH amino acid sequences within a family (bacteria, yeast, plants) is over 80%, while the homology of SauFDH with other bacterial FDHs (including enzymes from other pathogenic bacteria) is only 40% [2, 3].
Earlier, we cloned the saufdh gene into the pET23a plasmid and expressed active recombinant SauFDH in E. coli cells [5]. In this study, we developed an optimized procedure for the enzyme purification, which allowed to obtain SauFDH in amounts sufficient for establishment the optimal conditions for its crystallization and to get preliminary data on the SauFDH structure both in a free form and in a complex with the coenzyme (apo- and holoenzymes, respectively).
MATERIALS AND METHODS
SauFDH expression in E. coli cells. E. coli BL21(DE3) CodonPlus/pLysS cells were transformed with the pSauFDH plasmid generated by cloning of the saufdh gene into the pET23a plasmid. A single colony of the transformed cells was taked for expression and placed in 4 ml of 2YT medium [16 g/liter bactotrypton, 10 g/liter yeast extract (Difco, USA), NaCl 5 g/liter, pH 7.0] containing 25 µg/ml chloramphenicol and 150 µg/ml ampicillin and grown on a rotating shaker at 180 rpm at 37°C for 12 h. The overnight culture (60 µl) was transferred into a baffled-bottom 500-ml shaked flask with 60 ml of 2YT medium containing 25 µg/ml chloramphenicol and 150 µg/ml ampicillin and cultivated on a shaker at 120 rpm at 37°C. When the absorbance of the medium at 600 nm (A600) reached 0.6-0.8, the content of the flask was divided into three equal 20-ml portions and each portion was transferred into 1-liter baffled-bottom flask containing 180 ml of 2YT medium without antibiotics and cultivated at 30°C and 120 rpm. When the absorbance (A600) reached 0.6-0.8, protein synthesis was induced by adding lactose solution (300 g/liter) to the final concentration of 20 g/liter, the temperature was decreased to 20°C, and the cells were cultivated overnight (20°C, 120 rpm). Next morning, the cells were collected by centrifugation in a Beckman J-21 centrifuge (Beckman, USA) for 20 min at 5000 rpm at 4°C (J-14 rotor).
SauFDH purification. The produced cell biomass was resuspended in 0.1 M Na-phosphate buffer, pH 8.0 (cell concentration, 10 wt %) and frozen at –20°C. After thawing, the cells were disintegrated by sonication with a Branson Ultrasonic 250 cell disintegrator (Branson, USA); the lysate was heated at 55°C for 20 min and then incubated at room temperature for 30 min. The cell debris was separated by centrifugation (30 min, 18,000 rpm, 4°C, rotor J-20) and discarded. Solid ammonium sulfate was added to the supernatant (35% saturation), followed by incubation at 4°C for 4-4.5 h. The resulting precipitate was removed by centrifugation (30 min, 18,000 rpm, 4°C, J-20 rotor); the supernatant was collected and its volume was measured. Solid ammonium sulfate was added carefully to the supernatant to 85% of saturation with mixing. The produced solution was kept overnight at 4°C and then centrifuged for 30 min (18,000 rpm, 4°C, rotor J-20). The supernatant was discarded, and ammonium sulfate solution in 0.1 M Na-phosphate buffer, pH 7.0 (35% of saturation; solution A) was added to and carefully mixed with the pellet and incubated at 4°C for 1.5-2 h. Insoluble proteins were separated by centrifugation (30 min, 16,000 rpm, 4°C, rotor J-20), and the enzyme solution was loaded onto a high-substitution Phenyl Sepharose FF column (2.5 × 12 cm; Pharmacia Biotech, Austria) equilibrated with solution A. The column was washed with solution A until no absorption at 280 nm was observed in the eluate, and SauFDH was eluted with a descending linear gradient of ammonium sulfate concentration (35-0% saturation) in 0.1 M phosphate buffer, pH 7.0 (total eluent volume, 500 ml). The eluted fractions (5 ml) were assayed for the protein content by measuring absorbance at 280 nm (A280) and enzymatic activity (A). Fractions with the same activity/A280 ratios were used in further experiments. The enzyme was desalted and transferred into a required buffer by gel filtration through Sephadex G25 (Pharmacia Fine Chemicals, Sweden).
Solubility of SauFDH in ammonium sulfate solutions. To determine the dependence of SauFDH solubility on the (NH4)2SO4 concentration, 0.6-1.8 ml of saturated ammonium sulfate solution in 0.1 M Na-phosphate buffer, pH 7.0, was added to 100 µl of the cell-free extract (20 U/ml), and the volume of the sample was adjusted to 2 ml with the buffer. The samples were incubated at room temperature for 5 h, followed by centrifugation in an Eppendorf 5415D centrifuge (Eppendorf, Germany) for 15 min at 14,000 rpm at 4°C. The obtained solutions (25-µl aliquots) were tested for the residual enzyme activity.
SauFDH binding to the hydrophobic sorbent. The enzyme solution (solution B) prepared by dissolving precipitated enzyme in solution A (ammonium sulfate at 35% saturation in 0.1 M Na-phosphate buffer, pH 7.0) was used for determining the optimal concentration of ammonium sulfate for loading on a Phenyl Sepharose FF column. SauFDH solutions with different ammonium sulfate concentrations (5-35% saturation) were prepared by adding certain volumes of ammonium sulfate (35% saturation) and phosphate buffer to 200 µl of solution B to a total volume of 1.6 ml. An aliquot (400 µl) of 50% suspension of Phenyl Sepharose Fast Flow in ammonium sulfate solution of the required concentration was added to the samples. The samples were thoroughly mixed and left on the bench for 15 min to allow the resin to precipitate. Next, 25-µl aliquots of the supernatant were tested for the residual enzyme activity.
Enzyme activity assay. The activity of SauFDH was determined at 30°C from the accumulation of NADH in 0.1 M sodium-phosphate buffer, pH 7.0, based on absorbance at 340 nm (ε340 = 6220 M–1·cm–1) using a Schimadzu UV 1800 spectrophotometer. The concentrations of sodium formate and NAD+ in the reaction mixture were 0.6 M and 1.5 mg/ml, respectively.
The purity of SauFDH preparations at different purification stages was evaluated by analytical electrophoresis in 12% polyacrylamide gel in the presence of 0.1% sodium dodecyl sulfate using a Mini Protean II system (Bio-Rad, USA) according to the procedures recommended by the manufacturer.
SauFDH concentration in the preparation for crystallization was determined from the absorbance at 280 nm with a Shimadzu UV 1800 spectrophotometer (Germany) using the molar extinction coefficient for dimeric SauFDH ε280 = 57180 M–1·cm–1, which was calculated according to the equation suggested in [6]: ε280 = 5690·NTrp + 1280·NTyr, where NTrp and NTyr are the numbers of Trp and Tyr residues in the enzyme molecule (six and nine residues, respectively, in SauFDH).
The SauFDH concentration was converted from molar concentration to the mg/ml by dividing the molar concentration by the enzyme dimer molecular mass (Mr, 75,973.28 Da).
The total protein concentration at different purification stages was determined with the Bradford protein assay. Recombinant FDH from Pseudomonas sp. 101 (PseFDH) was used as a standard. The difference between the concentrations of purified SauFDH determined from the UV absorbance and by the Bradford method was less than 10%.
Analysis of SauFDH preparations by MALDI-TOF/TOF mass spectrometry. The experiments were conducted using equipment of the Industrial Biotechnology Center for Collective Use, Fundamentals of Biotechnology Federal Research Center, Russian Academy of Sciences.
The samples for mass spectrometry were prepared by tryptic hydrolysis of SauFDH in the polyacrylamide gel. A fragment of the gel (3-4 mm3) containing the enzyme band was cut from the Coomassie Brilliant Blue-stained gel (Fig. 1) and washed twice with 100 µl of 40% acetonitrile in 0.1 M NH4HCO3 for 20 min at 37°C to remove the dye. Next, 100 µl of acetonitrile was added to the gel sample for dehydration. Acetonitrile was then removed by drying, and 3.5 µl of modified trypsin solution (15 µg/ml, Promega) in 0.05 M NH4HCO3 was added to the sample. Hydrolysis was carried out for 20 h at 37°C; 5.25 µl of 0.5% trifluoroacetic acid (TFA) solution in 50% aqueous acetonitrile was then added and thoroughly mixed. The resulting supernatant was analyzed by MALDI-TOF/TOF mass spectrometry.
Fig. 1. Analytical SDS-PAGE of SauFDH preparations at different purification stages: 1) cell lysate after sonication, 2) after thermal treatment and ammonium sulfate fractionation, 3-7) fractions after hydrophobic chromatography, 8) after desalting; M, molecular mass markers, kDa.

Sample aliquots (1.5 µl) and 0.5 µl of 10 mg/ml 2,5-dihydroxybenzoic acid in 20% aqueous acetonitrile, 0.5% TFA (Aldrich, USA) were mixed on a target and air-dried.
Mass spectra were recorded with a MALDI-TOF/TOF Ultraflextreme BRUKER mass spectrometer (Germany) equipped with a UV laser (Nd) in the positive-ion reflectron mode. The accuracy of the monoisotopic mass measurement after calibration using peaks of trypsin autolysate was 0.002-0.011% (20-110 ppm). The spectra were recorded in the mass range m/z 500-6500 by adjusting the laser power to produce the best resolution.
Proteins were identified using the Mascot program (www.matrixscience.com). Mass spectra were processed with the FlexAnalysis 3.3 software package (Bruker Daltonics, Germany). The local database was search with the above accuracy using the Mascot program (peptide fingerprinting option) considering the following possible modifications: acetyl (protein N-terminus), Gln→pyroGlu (N-terminal Q), oxidation (M), and propionamide (C). Protein candidates with the reliability score >42 in the NCBI database were considered as reliably identified (p < 0.05).
Crystallization. The conditions for SauFDH crystallization were first optimized using the vapor diffusion technique (sitting drop version) with a Rigaku robotic crystallization system (USA) [7] using 10 mg/ml protein preparations in 0.1 M Tris-HCl (pH 8.0) and a standard kits for crystallization of globular proteins from Hampton Research (USA): Crystal Screen HT, Crystal Screen Cryo HT, Index HT, PEG/Ion HT, PEGRx HT, and SaltRx HT. Crystallization was done in 96-well crystallization plates (ArtRobbins).
The SauFDH holoenzyme was prepared by adding NAD+ and formate competitive inhibitor sodium azide to final concentrations of 7.0 and 0.11 mM, respectively, to the enzyme preparation prior to crystallization.
The selected crystallization conditions were further optimized at 15°C using the vapor diffusion technique (hanging drop version) in 24-well plates (VDX, USA). Each well contained 400 µl of precipitating solution, 1.5 µl of protein solution, and an equivalent amount of reservoir solution, which were mixed and applied onto a siliconized 22-mm glass (Hampton Research, USA).
Collection and processing of X-ray diffraction data. Prior to collecting X-ray diffraction data, protein crystals were withdrawn from the crystallization mixture with a loop and transferred to the cryosolution containing 25% glycerol and components of the reservoir solution. Next, the crystal in the loop was frozen in nitrogen vapor. Diffraction datasets were collected at 100 K using the synchrotron X-ray source Spring8 (station BL41XU) and European synchrotron radiation facilities (station ID29 [8]). The strategy for data collection was calculated with the HKL2000 [9] and BEST [10] programs. The datasets were processed using the Mosflm [11] and XDS [12] programs.
RESULTS AND DISCUSSION
Recombinant SauFDH. Preparation of protein crystals is the vital step in determining protein structure by X-ray diffraction analysis. The success of crystallization, in turn, depends on the purity and quality of the enzyme preparation. Metal-chelate affinity chromatography is often used for producing highly purified proteins tagged with 6 to 12 histidine residues (His-tag) at the N- or C-termini. However, the presence of the tag often changes enzyme’s properties, as it has been demonstrated for FDHs from Ogataea parapolymorpha DL-1 [5, 13], Candida methylica [14], Chaetomium thermophilum [15], and Pseudomonas sp. 101 (PseFDH) [16]. The changes in the catalytic properties and stability of the enzymes were likely associated with the effect of His-tag on the enzyme structure. Considering that the SauFDH structure may be used for the structure-based search of selective inhibitors for the native enzyme, we decided to produce the enzyme without affinity tags for crystallization.
Cultivation of SauFDH-producing strain. Earlier, our laboratory has developed a technology for producing recombinant FDHs from both prokaryotes and eukaryotes using an expression system based on the pET series plasmids and E. coli BL21(DE3) cells [2, 5]. In this system, the amount of expressed FDH is up to 40% of total soluble cell protein. Such a high content of the target enzyme allows obtaining virtually homogenous preparation in a minimal number of purification steps without introduction of affinity tags. Here, we used this system for SauFDH production. Similarly to other FDHs, the recombinant strain produced SauFDH in very high amounts. Analytical electrophoresis of the cell-free extract of E. coli cells expressing SauFDH (Fig. 1, lane 1) demonstrates that the content of produced SauFDH was no less than 35-40% of total soluble protein. The yield of SauFDH at the cultivation step was ~600 mg of the enzyme per 600 ml of culture fluid (1 g per liter of medium), which was very high for the cultivation of cells in shaking flasks.
Purification of recombinant SauFDH. To produce highly purified SauFDH preparations, we used a technique developed for the purification of FDHs expressed in E. coli cells, which is based on the fractionation of cell-free extract with ammonium sulfate followed by hydrophobic chromatography on Phenyl Sepharose FF [17]. In the case of FDHs with a high thermal stability, the cell-free extract is treated at 55°C for 15-25 min prior to fractionation with (NH4)2SO4 [2]. The data of differential scanning calorimetry showed that the thermal stability of SauFDH is very close to that of PseFDH [18], which up to now had been believed to have the highest thermal stability among FDHs. That is why we introduced thermal treatment of the cell-free extract for 20 min at 55°C as a step in SauFDH purification. Such treatment results in denaturation of some E. coli proteins with the production of well-formed precipitate, which simplifies the following fractionation with ammonium sulfate.
Ammonium sulfate fractionation required a separate set of experiments to determine the optimal (NH4)2SO4 concentrations for the enzyme reprecipitation and binding to the hydrophobic carrier. The dependence of the SauFDH solubility on the ammonium sulfate concentration in solution is presented in Fig. 2a. Based on the obtained data, the concentrations of (NH4)2SO4 of 35 and 85% saturation were selected for removal of associated proteins and precipitation of the enzyme, respectively. The results of electrophoretic analysis of SauFDH preparation at different purification stages are shown in Fig. 1. The electrophoretic pattern in lane 2 demonstrates that thermal treatment and fractionation with ammonium sulfate increased the purity of the enzyme preparation to at least 80%.
Fig. 2. Effect of ammonium sulfate concentration on the solubility of SauFDH (a) and enzyme binding to Phenyl Sepharose FF (b); 0.1 M phosphate buffer, pH 7.0, 25°C.
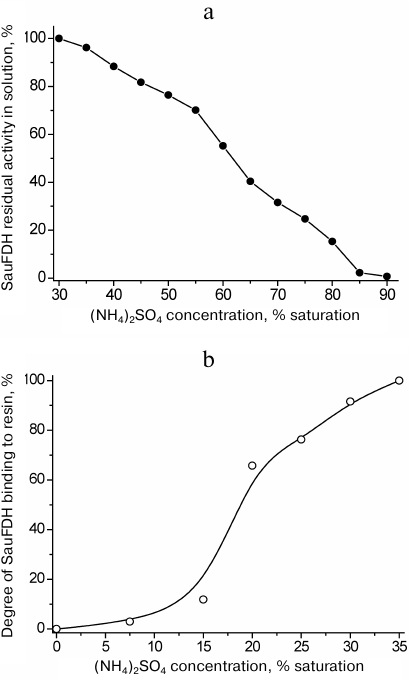
The final step of SauFDH purification from associated proteins was hydrophobic chromatography on Phenyl Sepharose Fast Flow. First, it was necessary to determine the optimal concentrations of ammonium sulfate for the enzyme binding to the sorbent and elution of associated proteins from the column prior to SauFDH desorption with a decreasing gradient of salt concentration. The efficiency of SauFDH absorption on Phenyl Sepharose FF at different ammonium sulfate concentration is shown in Fig. 2b. It can be seen from the figure that complete SauFDH binding to the carrier occurred only in 35% saturated (NH4)2SO4. Based on the data in Fig. 2 a and b, we used 35% saturated (NH4)2SO4 for SauFDH purification, which was different from the purification conditions used for other FDHs. In particular, 40 and 80% saturated ammonium sulfate was used for fractionation of PseFDH, while 30% saturated (NH4)2SO4 was used for loading this enzyme on a column with hydrophobic carrier and following washings [17]. The difference in behavior of these proteins in the presence of ammonium sulfate is likely related to the significant variations in their amino acid sequences (less than 40% homology [5]).
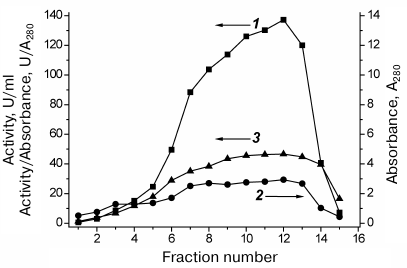
The results of SauFDH purification by hydrophobic chromatography are shown in Fig. 3. Express analysis of the enzyme purity in individual fractions was conducted based on the activity/A280 ratio, which was almost the same for fractions 7-13 (Fig. 3), suggesting similar SauFDH purity in these fractions. Analysis of fractions by denaturing electrophoresis (Fig. 1, lanes 3-7) demonstrated that the SauFDH preparations obtained by hydrophobic chromatography were virtually homogenous. In total, 400 mg of the highly purified enzyme was produced with the yield of 61% (based on enzyme activity).
Fig. 3. SauFDH purification by hydrophobic chromatography on Phenyl Sepharose FF in a descending gradient of ammonium sulfate concentration (35-0% saturation): 1) SauFDH activity in individual fractions, U/ml; 2) absorbance at 280 nm (A280); 3) relative activity, U/A280. Axes corresponding to particular curves are indicated with arrows; 0.1 M Na-phosphate buffer, pH 7.0.
However, calculations of the specific activity of purified SauFDH produced an unexpected result – the specific activity of the enzyme was 20 U/mg protein. The highest activity previously reported for bacterial FDHs was 10 U/mg. The maximal specific activity of FDHs from yeast and plants was 6.5 U/mg protein [2, 3, 5, 19, 20]. The use of rational protein design to enhance the catalytic properties of the bacterial enzymes resulted in improvement of the KM values for formate and NAD+ coenzyme, but not the catalytic constants [20, 21]. The specific activities of FDHs from Candida boidinii yeast and soya were increased 1.7-fold using directed evolution and rational protein design approaches, respectively [22, 23]. Hence, SauFDH is at least twice more active than any other known FDH. FDHs are commonly used for NAD(P)H regeneration in the chiral synthesis catalyzed by oxidoreductases [2, 5], which makes SauFDH a very promising enzyme for practical applications. Determination of the 3D structure of SauFDH might help in elucidating the causes for such high enzymatic activity of this enzyme.
Analysis of the obtained SauFDH preparation by MALDI-TOF/TOF mass spectrometry demonstrated the identity of its amino acid sequence to the one encoded by the saufdh2 gene in the pET23a plasmid; hence, the enzyme was not subjected to post-translational modifications during expression and purification.
Production of homogenous SauFDH in large quantities allowed us to crystallize it. SauFDH is a very stable enzyme [18] that retains its activity for 12 months during storage at 4°C. One batch of the produced SauFDH preparation was sufficient for conducting the entire cycle of crystallization experiments.
Crystallization. The screening of crystallization conditions for the free SauFDH and its complex with the cofactor was carried out in 96-well crystallization plates (ArtRobbins) with each well containing three smaller wells for different protein concentration under the same crystallization conditions. One crystallization plate allowed screening of 96 × 3 = 288 crystallization conditions. In total, six plates were prepared that were tested at 15 and 4°C (1728 variants in total). The ratios between the protein and reservoir solution in the small wells were 1 : 1, 2 : 1, and 1 : 2. The total volume of the protein solution in the small well was 0.1 µl for the ratios 1 : 1 and 2 : 1 and 0.2 µl for the ratio 1 : 2. The volume of the reservoir solution was 50 µl.
The first crystals of the apoenzyme were produced under the following conditions: 0.1 M HEPES, pH 7.5, 2% PEG 400, 2 M ammonium sulfate. The crystals were grown at 15°C for 7 days; they have a rhomboid shape with the length of the longest side of approximately 30 µm. Unfortunately, the size of the crystals was too small and required further optimization of the crystallization conditions.
NAD+ and sodium azide were added to the protein solution in order to obtain crystals of the holoenzyme. Next, the solution was centrifuged to remove impurities and possible aggregates and subjected to preliminary screening using the procedure described above. As a result of this screening, two sets of conditions were identified for the SauFDH holoenzyme crystallization: variant 1 – 0.2 M CaCl2, 0.1 M sodium acetate, pH 4.6, 30% 2-methylpentadiol-2,4; and variant 2 – 0.1 M HEPES-Na, pH 7.5, 1.4 M trisodium citrate. Under both conditions, the protein formed microcrystalline deposits that could not be used for X-ray diffraction analysis due to their small size (~10 µm) and inadequate morphology.
Considering that crystals produced for both forms of the enzyme were unsuitable for the X-ray diffraction analysis, we performed another round of the optimization of crystallization conditions that involved varying crystallization parameters in a narrow range around the ones identified in the first round. The crystals of the apoenzyme suitable for the X-ray diffraction analysis were produced under the following conditions: 0.1 M HEPES, pH 7.0, 2% PEG 400, 0.1 M sodium chloride, and 1.9 M ammonium sulfate (Fig. 4a). The crystals of the SauFDH holoenzyme were produced within 30 days at 15°C in 0.1 M HEPES, pH 7.5, 1.4 M trisodium citrate, and 10% trehalose (Fig. 4b). As seen in Fig. 4, a and b, the quality of the apoenzyme crystals was better than the quality of the holoenzyme crystals (for comparison, the image of the holoenzyme crystals is magnified; the actual sizes of the apo- and holoenzyme crystals were 200 and 40 µm, respectively). Nevertheless, the quality of the obtained crystals was sufficient for their examination by the X-ray diffraction analysis.
Fig. 4. Crystals of the SauFDH apo- (a) and holoenzyme (b) grown under the optimized conditions.

Collection and processing of the X-ray diffraction data. X-ray diffraction datasets were collected at 100 K using the Spring8 (station BL41XU) and European synchrotron radiation facilities (station ID29) as synchrotron sources (Table 1).
Crystallography data and parameters of SauFDH apo- and holoenzyme
crystals
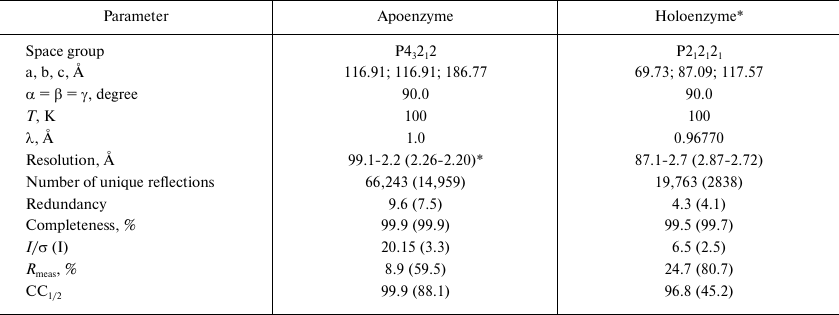
* Crystallization in the presence of 7.0 mM NAD+ and 0.11 mM
sodium azide.
** Data in parentheses are for the highest resolution shell.
The produced crystals of the SauFDH apo- and holoenzymes diffracted at the resolution of 2.2 and 2.7 Å, respectively, and belonged to different space groups (Table), which might be related to the cofactor binding in the holoenzyme. Estimation of the solvent content in the unit cell performed according to the procedure suggested by Matthews [24] using the CCP4i software package [25] showed that the independent unit contained two protein subunits of either apo- or holoenzyme, which is typical for this class of enzymes functioning as stable homodimers.
In conclusion, we prepared a new recombinant FDH from the pathogenic bacterium S. aureus that demonstrated the highest specific activity among the previously reported FDHs and obtained the crystals of this enzyme suitable for producing X-ray diffraction data. The data on the 3D structure of the SauFDH apo- and holoenzymes might be useful in the search for highly efficient enzyme inhibitors.
Acknowledgments. Some experiments were carried out using equipment from the Industrial Biotechnology Center of Collective Use, Fundamentals of Biotechnology Research Centre, Russian Academy of Sciences.
Funding. This work was supported by the Russian Foundation for Basic Research (projects Nos. 17-04-01662 and 20-04-00915, production and crystallization of enzymes), by the Federal Space Agency of Russia (Crystallizer experiment, crystallization and collection of X-ray diffraction data), and by the Ministry of Science and Higher Education of the Russian Federation (preliminary analysis of X-ray diffraction data).
Conflict of interest. The authors declare no conflict of interest.
Ethical approval. This article does not contain any studies with humans or animals performed by any of the authors.
REFERENCES
1.Tishkov, V. I., and Popov, V. O. (2004) Catalytic
mechanism and application of formate dehydrogenase, Biochemistry
(Moscow), 69, 1252-1267,
doi: 10.1007/s10541-005-0071-x.
2.Tishkov, V. I., and Popov, V. O. (2006) Protein
engineering of formate dehydrogenase, Biomol. Eng., 23,
89-110, doi: 10.1016/j.bioeng.2006.02.003.
3.Alekseeva, A. A., Savin, S. S., and Tishkov, V. I.
(2011) NAD+-dependent formate dehydrogenase from plants,
Acta Naturae, 3, 38-54, PMID: 22649703.
4.Resch, A., Rosenstein, R., Nerz, C., and Gotz, F.
(2005) Differential gene expression profiling of Staphylococcus
aureus cultivated under biofilm and planktonic conditions, Appl.
Environ. Microbiol., 71, 2663-2676.
5.Tishkov, V. I., Pometun, A. A., Stepashkina, A. V.,
Fedorchuk, V. V., Zarubina, S. A., Kargov, I. S., Atroshenko, D. L.,
Parshin, P. D., Kovalevski, R. P., Boiko, K. M., Eldarov, M. A.,
D’Oronzo, E., Facheris, S., Secundo, F., and Savin, S. S. (2018)
Rational design of practically important enzymes, Moscow Univ.
Chem. Bull., 73, 1-6, doi:
10.3103/S0027131418020153.
6.Pace, C. N., Vajdos, F., Fee, L., Grimsley, G., and
Gray, T. (1995) How to measure and predict the molar absorption
coefficient of a protein, Protein Sci., 4, 2411-2423,
doi: 10.1002/pro.5560041120.
7.Boyko, K. M., Lipkin, A. V., Popov, V. O., and
Kovalchuk, M. V. (2013) From gene to structure: the protein factory of
the NBICS centre of Kurchatov institute, Crystallogr. Rep.,
58, 442-449, doi: 10.1134/S106377451105004x.
8.De Sanctis, D., Beteva, A., Caserotto, H., Dobias,
F., Gabadinho, J., Giraud, T., Gobbo, A., Guijarro, M., Lentini, M.,
Lavault, B., Mairs, T., McSweeney, S., Petitdemange, S., Rey-Bakaikoa,
V., Surr, J., Theveneau, P., Leonard, G. A., and Mueller-Dieckmann, C.
(2012) ID29: a high-intensity highly automated ESRF beamline for
macromolecular crystallography experiments exploiting anomalous
scattering, J. Synchrotron Radiat., 19, 455-461,
doi: 10.1107/S0909049512009715.
9.Otwinowski, Z., and Minor, W. (1997) Processing of
X-ray diffraction data collected in oscillation mode, Methods
Enzymol., 276, 307-326.
10.Bourenkov, G. P., and Popov, A. N. (2006) A
quantitative approach to data-collection strategies, Acta
Crystallogr. D Biol. Crystallogr., 62, 58-64,
doi: 10.1107/S0907444905033998.
11.Battye, T. G. G., Kontogiannis, L., Johnson, O.,
Powell, H. R., and Leslie, A. G. W. (2011) iMOSFLM: a new graphical
interface for diffraction-image processing with MOSFLM, Acta
Crystallogr. D Biol. Crystallogr., 67, 271-281,
doi: 10.1107/S0907444910048675.
12.Kabsch, W. (2010) XDS, Acta Crystallogr. D
Biol. Crystallogr., 66, 125-132,
doi: 10.1107/S0907444909047337.
13.Yu, S., Zhu, L., Zhou, C., An, T., Zhang, T.,
Jiang, B., and Mu, W. (2014) Promising properties of a formate
dehydrogenase from a methanol-assimilating yeast Ogataea
parapolymorpha DL-1 in His-tagged form, Appl. Microbiol.
Biotechnol., 98, 1621-1630,
doi: 10.1007/s00253-013-4996-5.
14.Ordu, E. B., and Karagüler, N. G. (2007)
Improving the purification of NAD+-dependent formate
dehydrogenase from Candida methylica, Prepar. Biochem.
Biotechnol., 37, 333-341,
doi: 10.1080/10826060701593233.
15.Esen, H., Alpdağtaş, S., Mervan
Çakar, M., and Binay, B. (2019) Tailoring of recombinant FDH:
effect of histidine tag location on solubility and catalytic properties
of Chaetomium thermophilum formate dehydrogenase (CtFDH),
Prepar. Biochem. Biotechnol., 49, 529-534,
doi: 10.1080/10826068.2019.1599394.
16.Pometun, A. A., Parshin, P. D., Galanicheva, N.
P., Uporov, I. V., Atroshenko, D. L., Savin, S. S., and Tishkov, V. I.
(2020) Effect of His6 sequence on properties of formate
dehydrogenases from bacterium Pseudomonas sp. 101, Moscow
Univ. Chem. Bull., 75, 4.
17.Rojkova, A. M., Galkin, A. G., Kulakova, L. B.,
Serov, A. E., Savitsky, P. A., Fedorchuk, V. V., and Tishkov, V. I.
(1999) Bacterial formate dehydrogenase. Increasing the enzyme thermal
stability by hydrophobization of alpha helices, FEBS Lett.,
445, 183-188, doi: 10.1016/S0014-5793(99)00127-1.
18.Pometun, A. A., Kleymenov, S. Yu., Zarubina, S.
A., Kargov, I. S., Parshin, P. D., Sadykhov, E. G., Savin, S. S., and
Tishkov, V. I. (2018) Comparison of thermal stability of new formate
dehydrogenases with differential scanning calorimetry, Moscow Univ.
Chem. Bull., 73, 80-84,
doi: 10.3103/S002713141802013X.
19.Slusarczyk, H., Felber, S., Kula, M. R., and
Pohl, M. (2000) Stabilization of NAD-dependent formate dehydrogenase
from Candida boidinii by site-directed mutagenesis of cysteine
residues, Eur. J. Biochem., 267, 1280-1289,
doi: 10.1046/j.1432-1327.2000.01123.x.
20.Tishkov, V. I., Goncharenko, K. V., Alekseeva, A.
A., Kleymenov, S. Yu., and Savin, S. S. (2015) Role of a structurally
equivalent phenylalanine residue in catalysis and thermal stability of
formate dehydrogenases from different sources, Biochemistry
(Moscow), 80, 1690-1700,
doi: 10.1134/S000629791513005.
21.Alekseeva, A. A., Fedorchuk, V. V., Zarubina, S.
A., Sadykhov, E. G., Matorin, A. D., Savin, S. S., and Tishkov, V. I.
(2015) Role of Ala198 in stability and coenzyme specificity of
bacterial formate dehydrogenases, Acta Naturae, 7, 60-69,
PMID: 25927002.
22.Slusarczyk, H., Felber, S., Kula, M.-R., and
Pohl, M. (2003) Novel mutants of formate dehydrogenase from Candida
boidinii, US Patent Application Publication, US2003/0157664,
21.09.2003.
23.Kargov, I. S., Kleymenov, S. Y., Savin, S. S.,
Tishkov, V. I., and Alekseeva, A. A. (2015) Improvement of the soy
formate dehydrogenase properties by rational design, Prot. Eng.
Des. Select., 28, 171-178,
doi: 10.1093/protein/gzv007.
24.Matthews, B. W. (1968) Solvent content of protein
crystals, J. Mol. Biol., 33, 491-497,
doi: 10.1016/0022-2836(68)90205-2.
25.Winn, M. D., Ballard, C. C., Cowtan, K. D.,
Dodson, E. J., Emsley, P., Evans, P. R., Keegan, R. M., Krissinel, E.
B., Leslie, A. G., McCoy, A., McNicholas, S. J., Murshudov, G. N.,
Pannu, N. S., Potterton, E. A., Powell, H. R., Read, R. J., Vagin, A.,
and Wilson, K. S. (2011) Overview of the CCP4 suite and current
developments, Acta Crystallogr. D Biol. Crystallogr., 67,
235-242, doi: 10.1107/S0907444910045749.