Role of Interactions of the CRE Region of Escherichia coli RNA Polymerase with Nontemplate DNA during Promoter Escape
I. V. Petushkov1a* and A. V. Kulbachinskiy1
1Institute of Molecular Genetics, Russian Academy of Sciences, 123182 Moscow, Russia* To whom correspondence should be addressed.
Received May 7, 2020; Revised June 9, 2020; Accepted June 10, 2020
RNA polymerase (RNAP) recognizes promoter DNA through many interactions that determine specificity of transcription initiation. In addition to the dedicated transcription initiation σ factor in bacteria, the core enzyme of RNAP can also participate in promoter recognition. In particular, guanine residue at the +2 position (+2G) of the nontemplate DNA strand is bound in the CRE pocket formed by the RNAP β subunit. Here, we analyzed the role of these contacts in the process of promoter escape by RNAP by studying point mutations in the β subunit of Escherichia coli RNAP that disrupted these interactions. We found that the presence of +2G in the promoter slowed down the rate of promoter escape and increased proportion of inactive complexes. Amino acid substitutions in the CRE pocket decreased the promoter complex stability and changed the pattern of short RNA products synthesized during initiation, but did not significantly affect the rate of transition to elongation, regardless of the presence of +2G. Thus, the contacts of the CRE pocket with +2G do not make a significant contribution to the kinetics of promoter escape by RNAP, while the observed changes in the efficiency of abortive synthesis are not directly related to the rate of promoter escape.
KEY WORDS: RNA polymerase, CRE pocket, promoter escapeDOI: 10.1134/S000629792007007X
Abbreviations: CRE, core recognition element; ITS, initially transcribed sequence; RNAP, RNA polymerase; RPint, initiating RNAP-promoter complex; RPo, open RNAP-promoter complex; WT, wild type.
INTRODUCTION
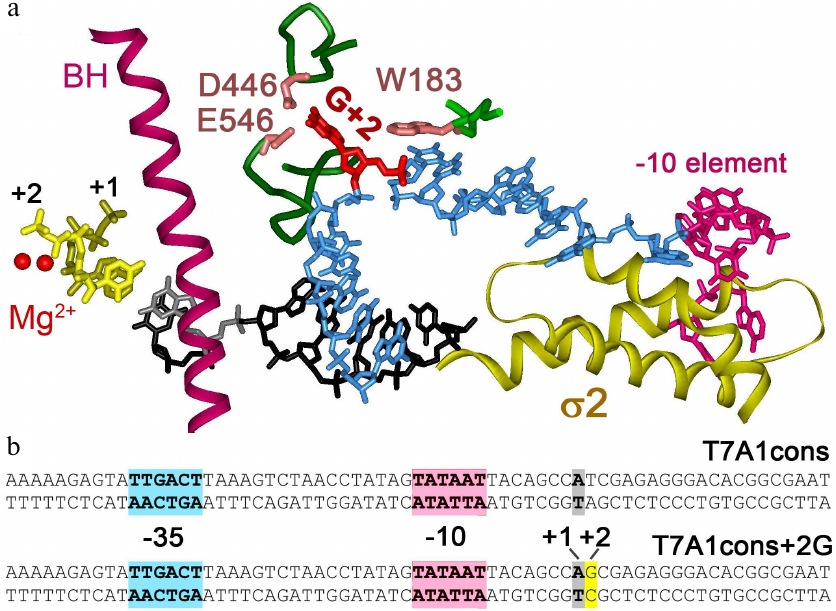
To initiate transcription, RNA polymerase (RNAP) locally melts ~13 base pairs of DNA duplex at the transcription start point, resulting in the formation of an open promoter complex (RPo) [1]. During promoter binding, the –35, –10 elements of the promoter and the discriminator region downstream of the –10 element are recognized by the σ factor. The β subunit of the RNAP core enzyme can also form specific contacts with nucleotides of the nontemplate DNA strand in the melted region from –4 to +2 positions, which is called core recognition element (CRE). Interactions of the guanine residue at the position +2 (+2G) are of particular interest. This residue is unstacked from the surrounding nucleotides and is placed in a special pocket formed by the conserved amino acid residues of the β subunit, the CRE pocket (Fig. 1a) [2-4]. Although these interactions were observed in structural studies of the promoter complex, analysis of the sequences of numerous promoters did not reveal any consensus motifs in this region suggesting that they are not strictly required for promoter recognition and may instead be important at later stages of transcription [5]. For example, in a number of promoters in E. coli their interactions with CRE pocket were shown to be important for determination of the transcription start site [6].
During the synthesis of the first 4-6 nucleotides of RNA transcript, the 5′-end of the growing RNA collides with the region σ 3.2, which directly contacts with the template DNA strand and positions it in the active center of RNAP [3, 7, 8]. The collision makes difficult further RNA extension and, as a result, causes transcriptional pausing at the +6 position [9]. However, pausing at this position could also be observed in the case of deletion of the region σ 3.2, because its formation is also facilitated by the TG motif located at the +6/+7 positions of the promoter [10]. Substitution D446A in the CRE pocket of the β subunit increases duration of the pause, indicating importance of the contacts of the CRE pocket with the guanine residue in the nontemplate strand for suppression of pausing during transcription initiation [10]. Furthermore, these contacts play a similar role in suppression of pauses at the elongation stage of transcription [11]. It has also been shown that mutations in the CRE pocket affect the overall rate of elongation and transcription termination efficiency. Thus, this region plays a role at all stages of transcription [12]. In addition to the CRE pocket, an adjacent conservative residue W183 in the β subunit, which interacts with the nucleotide residue at the +1 position of the nontemplate strand (Fig. 1), is also involved in the formation of promoter complexes. These contacts were shown to affect the efficiency of abortive RNA synthesis and also contribute to the formation of RPo on promoters of the alternative σ54 subunit [13].
Fig. 1. Interactions of Thermus thermophilus RNAP with nontemplate DNA strand in the transcription initiation complex [3]. a) Specific RNAP interactions with DNA near the active center. Two catalytic Mg2+ ions are indicated by red spheres. Bridge helix (BH) is shown in purple, first two RNA nucleotides are shown in yellow. Template DNA strand in the active center is shown in black, nontemplate DNA strand – in blue. Guanine residue at the +2 position (shown in red) is unstacked from DNA and interacts with CRE pocket of the β subunit (residues D446, E546 are shown). Nucleotide at the +1 position of the nontemplate strand interacts with the residue W183 in the β subunit. The –10 element of the promoter interacting with region 2 of the σA subunit is highlighted in pink. b) Promoter T7A1cons and its variant with the +2G substitution; the –35, –10 promoter elements and the transcription start point are shown.
During incorporation of the first nucleotides, which is accompanied by melting of the downstream DNA duplex, RNAP retains most contacts with promoter elements, resulting in the formation of a strained “scrunched” complex with a noncanonical size of the transcriptional bubble [14-16]. The accumulated stress can lead to a partial or complete backtracking of RNAP, leading to the release of an abortive RNA product. When the size of RNA transcript reaches a length of 9-15 nucleotides, the accumulated energy becomes enough to break contacts with the promoter, which contributes to the transition from the initiating complex (RPint) to the elongation complex [10, 16-20]. Direct interactions of the RNA transcript with the 3.2 region also make an important contribution to this process [9, 21-24].
Little is known about the kinetics of promoter escape and the factors that can modulate this process. It has been shown that stronger contacts with the key promoter elements slow down the rate of promoter escape [21, 25]. The effects of the initially transcribed sequence (ITS) on the kinetics of transcription initiation are more complex. Substitutions in the ITS can lead to up to 10-fold and 25-fold changes in the level of transcription in vivo and in vitro, respectively [26, 27]. Relative amounts of the abortive and full-length products, as well as the size of abortive products, depend on the ITS sequence [27-30]. Several factors were suggested to explain the observed effects of the ITS on the rate of promoter escape, including changes in the stability of the downstream DNA duplex, stability of the RNA-DNA heteroduplex, and proportion of the non-productive complexes formed during abortive synthesis [10, 31]. Such non-productive complexes were recently shown to form paused or partially backtracked states using single-molecule techniques [10, 32]. It can be assumed that specific contacts of the CRE nucleotides with the core enzyme of RNAP can affect kinetics of this process. By analyzing many ITS variants on different promoters, it was found that in the case of the lacUV5 promoter the +2G substitution increased the rate of transition to elongation, although the presence of additional contacts could have been expected to delay promoter clearance. This effect was specific for lacUV5, but was not observed for two other studied promoters [31]. Thus, the exact role of the CRE pocket and its interactions with +2G in the process of promoter escape remains unclear. Also, the role of interactions of the +1 nucleotide in the nontemplate strand with the residue W183 in this process has not been examined. In this study, we analyzed the roles of contacts of residues +1 and +2 in the nontemplate strand with the core enzyme of RNAP in the process of promoter escape, using a molecular beacon assay [33, 34] combined with transcription in vitro.
MATERIALS AND METHODS
Cloning and protein purification. RNAP core enzyme (with 6 histidine residues at the N-terminus of the β subunit) was expressed using the pIA679 plasmid encoding all RNAP subunits under the control of the T7 RNAP promoter [35]. Substitutions W183A, D446A, E546A in the β subunit were generated by site-directed mutagenesis in the rpoB gene [12]. The RNAP core enzyme was isolated from E. coli strain BL21(DE3). Protein expression, cell disruption, precipitation with Polymin P and ammonium sulfate (Sigma-Aldrich, USA) followed by chromatographic purification of the enzyme on heparin, Ni-affinity, and anion-exchange columns were carried out according to the published procedure [35]. Expression and chromatographic purification of the σ70 subunit was performed as previously described [7]. The σ70 subunit containing a unique cysteine residue at position 211 was expressed and purified by a similar procedure and labeled with tetramethylrhodamine-5-maleimide as described [34].
In vitro transcription. Transcription templates containing the T7A1cons promoter (from –85 to +53 positions relative to the start point) and its variant with the +2G substitution were generated by PCR using a Pfu DNA polymerase (Thermo Fisher Scientific, USA) and purified using a GeneJET PCR purification kit (Thermo Fisher Scientific) [12, 36]. The core enzyme (50 nM final concentration) and the σ70 subunit (250 nM) were mixed in a transcription buffer TB40 (40 mM Tris-HCl, Sigma-Aldrich), pH 7.9, 40 mM NaCl (Roth, Germany), 10 mM MgCl2 (Sigma-Aldrich), a T7A1cons template (25 nM) was added, and the samples were incubated for 10 minutes at 37°C. For detection of both abortive and full-length products, a mixture of ApU primer with final concentration 100 µM, ATP and CTP with final concentration 25 µM each, GTP – 7 µM and 2.5 µCi [α-32P]GTP were added. To obtain 5′-labeled abortive RNA transcripts, a mixture of GTP, CTP, UTP with final concentration 25 µM and 2.5 µCu [γ-32P]ATP was added. Transcription was stopped after 10 minutes by adding an equal volume of stop solution (8 M urea, 20 mM EDTA, Sigma-Aldrich). RNA products were analyzed using 23% or 30% denaturing PAGE; detection was performed using a Typhoon FLA 9500 scanner (GE Healthcare, USA).
Measurements of the promoter complex half-life and the rate of promoter escape by molecular beacon assay. All experiments were carried out in a TB40 buffer in the presence of 0.01% (v/v) Tween-20 in an 800 µl cuvette at 37°C. Fluorescence was excited at 550 nm and recorded at 580 nM (monochromator slit widths 10 nm) with a QuantaMaster QM40 fluorimeter (Photon Technology International, USA). The labeled σ70 subunit was added to the reaction mixture to final concentration of 1 nM. Then the core enzyme was added to final concentration of 4 nM and promoter DNA – to final concentration of 5 nM; next the mixture was incubated for 5 min. To measure stability of the promoter complex, heparin was added to concentration 100 µg/ml, the sample was mixed with a micropipette for about 10-15 seconds and the detector was switched on [37]. To measure the rate of promoter escape after RPo assembly, a 1000-fold excess of unlabeled σ70 subunit was added to the reaction mixture (to avoid reinitiation of transcription with labeled σ70) and then a mixture of all NTP was added to final concentration of 100 µM. Fluorescence intensity was normalized to the signal amplitude and approximated by the first order kinetics [25, 37].
RESULTS
The effect of point mutations in the CRE region on transcription from the T7A1cons promoter. In this study, we analyzed three mutant RNAP variants with substitutions D446A, E546A, and W183A in the β subunit, which were shown previously to disrupt interactions of RNAP with the +2 and +1 nucleotides of the nontemplate promoter strand [2, 12]. For this analysis, we used the T7A1cons promoter with the consensus –10 element (TATAAT) and a nearly consensus –35 element (TTGACT vs. TTGACA) (Fig. 1b). This promoter is highly active in the synthesis of full-length and abortive RNA products in vitro, forms stable complexes with RNAP and is therefore a good model for studying mechanisms of the promoter escape [3, 7, 8, 2, 38, 39]. We tested activity of the mutant polymerases on this promoter, as well as of its variant containing a guanine residue in the +2 position (+2G substituted for +2T, Fig. 1b).
To compare transcriptional activities of the wild-type and mutant RNAP variants, we first analyzed transcription in the presence of an incomplete set of nucleotide substrates, allowing for 20 nt RNA synthesis (since UTP, which should be included at the 21st position, was missing from the reaction mixture). It was found that under these conditions, all RNAPs were capable of transition to elongation and synthesized 20 nt RNA with comparable efficiencies (Fig. 2a). All polymerases also synthesized a large number of abortive products up to 15 nucleotides in length, but the efficiency of 6-14 nt RNA synthesis was lower for the D446A and E546A mutant RNAPs.
Fig. 2. Effect of substitutions in the CRE region of E. coli RNAP on RNA synthesis during transcription initiation. a) 20-Mer RNA synthesis from the T7A1cons promoter (23% denaturing PAGE). Profile of RNA products normalized to the intensity of the 20 nt product is shown on the left. b) RNA products obtained upon initiation of transcription from T7A1cons and T7A1cons+2G in the presence of [γ-32P]ATP (30% denaturing PAGE).
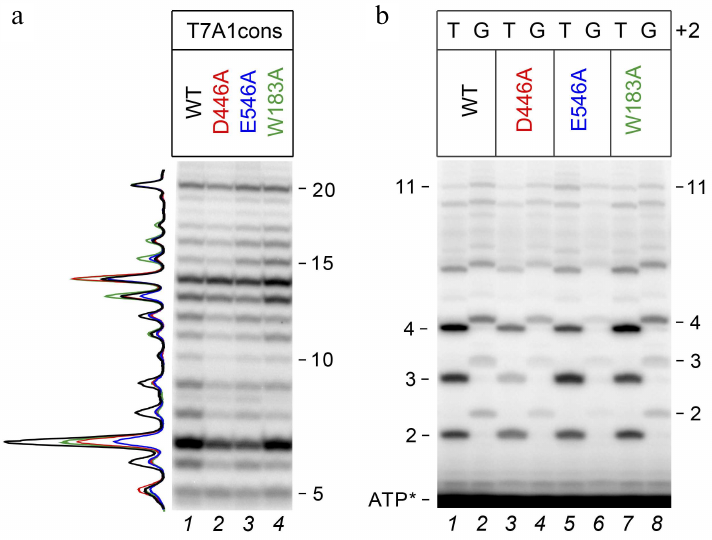
To assess the effects of mutations in RNAP and the +2G substitution on the synthesis of short abortive products, we performed transcription using [γ-32P]ATP, which allowed to label all synthesized RNAs at the 5′-end. It was shown that substitution +2G led to a noticeable decrease in the amounts of short RNAs with a length of 2-4 nt, but had a weaker effect on the synthesis of longer products (Fig. 2b). A similar effect was observed both in the case of wild-type RNAP (WT, compare lanes 1 and 2) and in the case of mutant RNAPs. In the case of mutant RNAPs with the D446A and E546A substitutions, an additional decrease in the amounts of 2-4 nt RNAs was observed for both promoter variants (compare lanes 1, 3, and 5 for T7A1cons and 2, 4, and 6 for T7A1cons+2G).
The ratio of abortive and productive RNA products is an important characteristic of the efficiency of transition from transcriptional initiation to elongation. Thus, the observed effects suggest that the +2G substitution in the promoter, as well as mutations in the CRE region of RNAP, could affect the rate of promoter escape by changing contacts of RNAP with the promoter DNA.
Measurements of stability of promoter complexes by molecular beacon assay. The molecular beacon assay is based on measurements of changes in the fluorescence of labeled σ factor upon binding of RNAP to promoter DNA. The fluorophore is covalently attached to the Cys residue at position 211 of the σ70 subunit located near the region 2, which recognizes the –10 promoter element. During formation of the promoter complex, DNA binding causes conformational changes of aromatic residues in the region 2 and removes them away from the fluorophore, which results in the increase of fluorescence. This allows real-time monitoring of the formation and dissociation of the promoter complex [33, 34, 40, 41].
To confirm the role of the +2G residue in stabilization of the promoter complex, we measured dissociation kinetics of the promoter complex in the presence of a DNA competitor heparin using the T7A1cons and T7A1cons+2G promoters. During complex dissociation, fluorescence decreases due to the loss of contacts between the σ subunit and the –10 promoter element (Fig. 3a). As expected, the +2G substitution increased stability of the promoter complex measured in this assay (half-life t1/2 increased from ~158 to ~209 s for WT RNAP; Fig. 3, a and b).
Fig. 3. Effect of the +2G residue on stability of the promoter complex. Measurements were carried out using the molecular beacon assay. a) Kinetics of the promoter complex dissociation in the presence of heparin for the T7A1cons and T7A1cons+2G promoters for WT and D446A RNAPs. b) Half-life of promoter complexes (t1/2) for WT and mutant RNAPs containing mutations in the CRE region on the studied promoters. Measurements with the W183A RNAP on the T7A1cons+2G promoter were not performed.
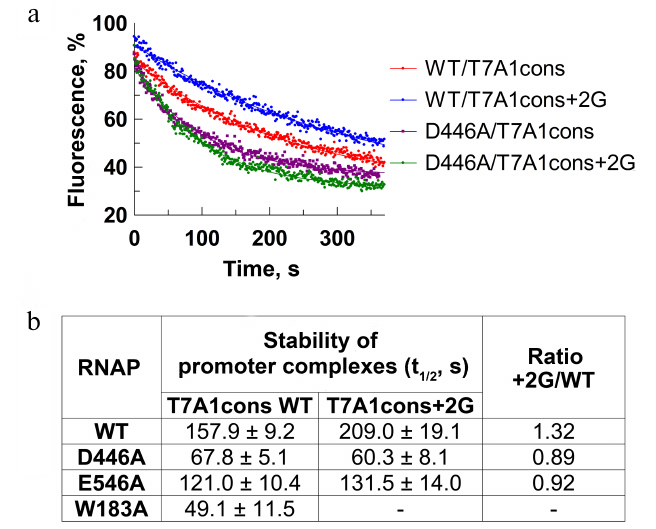
We also tested RNAPs with substitutions of amino acid residues that contact +2G [2, 6, 11, 12]. It was shown that the D446A substitution decreased stability of the T7A1cons promoter complex in comparison with the WT RNAP (t1/2 = 68 and 158 s, respectively; Fig. 3b). In addition, RNAP with this substitution was unable to recognize +2G in the promoter sequence (t1/2 was not increased in the case of T7A1cons+2G). The substitution E546A resulted in a similar, but weaker effect. The substitution W183A led to a significant destabilization of the promoter complex (Fig. 3b). Thus, in accordance with the published structural and biochemical data, the contacts of the +2G residue with the CRE pocket increased stability of the promoter complex demonstrating that this experimental system could be used to study the role of these interactions in promoter escape.
Measurements of the rate and efficiency of promoter escape by molecular beacon assay. Using the model system described above, we measured the rate and efficiency of RNAP escape from the T7A1cons and T7A1cons+2G promoters. Nucleotide substrates were added to the preformed promoter complexes and changes in fluorescence during the transition to transcription elongation were monitored in real time. The process of promoter escape can be described by the first-order kinetics (Fig. 4a). The time during which half of the active complexes escape from the promoter (t1/2) can be used as a measure of the rate of this process (Fig. 4b). Furthermore, the efficiency of promoter escape can be determined from the fraction of inactive complexes that do not break contacts with the promoter during the course of experiment, thus forming a plateau on the fluorescence curve (Fig. 4, a and c). This fraction could correspond to either catalytically inactive complexes or initiating complexes that are engaged in abortive RNA synthesis, but cannot leave the promoter. For WT RNAP, t1/2 was about 60 s, and the fraction of inactive promoter complex was about 8% (Fig. 4, a-c). The substitution +2G in the promoter led to a >2-fold decrease in the rate of promoter escape (t1/2 = 136 s), while the fraction of inactive complexes increased ~3-fold (up to 24%).
To understand whether the observed effects depend on the contacts of +2G with the CRE pocket, we tested the effects of the D446A and E546A substitutions on the transition to elongation. The substitution D446A did not affect the rate of escape from the T7A1cons promoter, but led to a significant increase in the proportion of inactive complexes (up to 30%). On the T7A1cons+2G promoter, the time of promoter escape for this RNAP increased to 101.5 s, while the proportion of inactive complexes reached 40%. The substitution E546A slightly increased the rate of escape from the T7A1cons promoter, but did not affect the fraction of inactive complexes. In the case of W183A RNAP, we observed a slight decrease of the rate of promoter clearance.
Fig. 4. Effect of residue +2G on the process of promoter escape. a) Kinetics of promoter escape for T7A1cons and T7A1cons+2G, measured for wild-type and D446A RNAPs. b) Promoter escape times (t1/2) for wild-type and mutant RNAPs with substitutions in the CRE region. c) Fractions of inactive initiation complex (RPint).
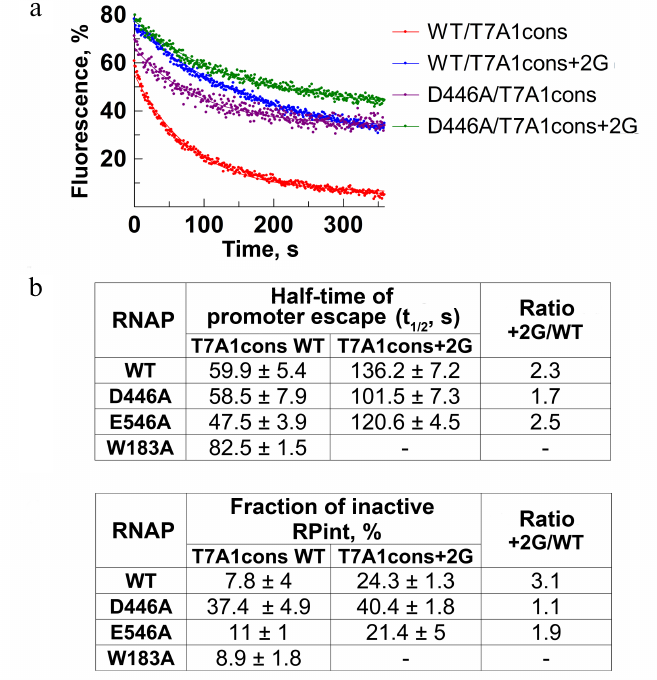
Thus, the results obtained by the molecular beacon assay indicate that the +2G substitution greatly reduces the rate of transition to elongation and increases the fraction of inactive complexes. Mutation D446A does not affect the rate of promoter escape but leads to the formation of a large amount of inactive promoter complexes.
DISCUSSION
In this work, we studied the role of RNAP contacts with the CRE region on stability of the promoter complexes and transition to elongation using the molecular beacon assay. This method allows detecting formation and breaking of the contacts of the σ factor with the –10 promoter element during transcription initiation. We showed that the presence of the +2G residue in the T7A1cons promoter increases stability of the promoter complexes, which was in agreement with the published data [2, 12]. Point amino acid substitutions D446A and E546A disrupt the interactions of the +2G residue with the CRE pocket and make promoter complexes insensitive to the +2G substitution. The substitution W183A leads to a significant decrease in the lifetime of the complexes (about 3-fold), which supports the available data on the role of interactions of W183 with the nontemplate promoter strand in stabilization of promoter complexes [12, 13]. The experiments confirm functional interactions of RNAP with the CRE region in this promoter.
This allowed us to study the role of these interactions in the process of promoter escape. It was shown that the point mutations in the CRE pocket of RNAP either do not affect the rate of promoter escape, or have a relatively small effect (a slight decrease in the rate is observed only for the W183A substitution). Furthermore, the presence of the +2G residue in the promoter leads to a significant decrease in the rate of promoter escape and an increase in the fraction of inactive complexes. This effect could be explained by the interaction of +2G with the CRE pocket. However, the D446A substitution can only partially suppress the effect of +2G in the T7A1cons+2G promoter (acceleration by ~25% relative to WT RNAP), while the E546A substitution has almost no effect on this process. Thus, the contacts of the CRE pocket with +2G are not critical for promoter escape. Based on the structural data for RPint from E. coli and T. thermophilus RNAPs containing short 4 and 6 nt RNA transcripts, interactions of +2G with the CRE pocket are not evident in the complexes [3, 4]. This indicates that these contacts are broken prior to the contacts of the nontemplate DNA strand with the σ subunit. This may likely explain why the substitution D446A has only a small effect on the rate of promoter escape.
Analysis of abortive RNA products in in vitro transcription experiments shows that the efficiency of abortive synthesis poorly correlates with the rate of promoter escape. Thus, the amount of short abortive transcripts synthesized on the T7A1cons+2G promoter is much smaller than in the case of T7A1cons lacking +2G. This is probably due to the fact that the presence of an additional rG/dC pair in the RNA-DNA hybrid (as compared with rU/dA in T7A1cons) results in a more stable retention of short RNAs in the initiation complex [42]. At the same time, the rate of transition to elongation in the case of this promoter is much lower. In addition, the proportion of inactive complexes is significantly higher in the case of the T7A1cons+2G promoter. This can be explained by a slower RNAP translocation or by formation of inactive transcription intermediates without dissociation of short RNA products in these promoter complexes. Indeed, single-molecule studies revealed transcriptional pausing during abortive synthesis, as well as temporal backtracking of the initiating complexes without transcript dissociation [10, 32]. The presence of such complexes has a significant effect on the rate of promoter escape [32]. Perhaps, the +2G substitution leads to the increase in the fraction of such complexes, which could be examined in further studies at the single-molecule level.
The D446A substitution has little effect on the rate of transition to elongation, although it decreases the efficiency of abortive synthesis. In addition, this mutation does not compensate for the decrease in the rate and efficiency of transition to elongation on the T7A1cons+2G promoter. This suggests that the effects observed on this promoter cannot be explained by the contacts of +2G with the CRE pocket. Moreover, the substitution D446A significantly increases the fraction of inactive complexes in the case of the T7A1cons promoter that does not contain +2G. It can be suggested that both specific (in the presence of +2G) and non-specific (in its absence) contacts of the residue D446 with the nontemplate DNA strand can suppress formation of inactive complexes during initiation. When this residue is mutated, a higher proportion of the complexes becomes inactive, but the active complexes leave the promoter at a rate comparable to the wild-type RNAP.
One can assume that disruption of the contacts of the RNAP holoenzyme with the promoter DNA should lead to acceleration of the rate of transition to elongation, but this is not true for the mutations in the CRE region. The substitution W183A slows down the rate of promoter clearance, although it decreases stability of the promoter complexes. It is likely that the contacts of W183 residue with nontemplate DNA strand could promote RNAP translocation during the initial stages of RNA synthesis. In the case of RNAP with the D446A substitution, there is also a decrease in stability of the promoter complexes, but there is no effect on the rate of promoter escape.
In conclusion, the presence of +2G residue in the initially transcribed region can affect not only stability of the promoter complexes but also transition to elongation, by changing efficiency of the abortive synthesis and the rate of RNAP escape from the promoter. Importantly, the intensity of the abortive RNA synthesis during initiation does not directly correlate with the rate of transition from initiation to elongation. Mutations in the CRE region of RNAP can increase the proportion of inactive initiating complexes (D446A RNAP) or decrease the rate of promoter escape (W183A RNAP). These effects are not related to the recognition of +2G, but could be explained by interactions of this region with subsequent nucleotides of the nontemplate DNA strand. Further studies of transcription initiation, primarily at the single-molecule level, can help to elucidate the detailed mechanisms of these processes.
Funding. This work was supported by the Russian Science Foundation (project No. 17-14-01393).
Acknowledgements. The authors are grateful to Dr. Irina Artsimovich for providing plasmids.
Ethics declarations. The authors declare no conflict of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
REFERENCES
1.Feklistov, A., Sharon, B. D., Darst, S. A., and
Gross, C. A. (2014) Bacterial sigma factors: a historical, structural,
and genomic perspective, Annu. Rev. Microbiol., 68,
357-376, doi: 10.1146/annurev-micro-092412-155737.
2.Zhang, Y., Feng, Y., Chatterjee, S., Tuske, S., Ho,
M. X., Arnold, E., and Ebright, R. H. (2012) Structural basis of
transcription initiation, Science, 338, 1076-1080,
doi: 10.1126/science.1227786.
3.Basu, R. S., Warner, B. A., Molodtsov, V., Pupov,
D., Esyunina, D., Fernandez-Tornero, C., Kulbachinskiy, A., and
Murakami, K. S. (2014) Structural basis of transcription initiation by
bacterial RNA polymerase holoenzyme, J. Biol. Chem., 289,
24549-24559, doi: 10.1074/jbc.M114.584037.
4.Zuo, Y., and Steitz, T. A. (2015) Crystal
structures of the E. coli transcription initiation complexes
with a complete bubble, Mol. Cell, 58, 534-540,
doi: 10.1016/j.molcel.2015.03.010.
5.Mitchell, J. E., Zheng, D., Busby, S. J., and
Minchin, S. D. (2003) Identification and analysis of
“extended –10” promoters in Escherichia
coli, Nucleic Acids Res., 31, 4689-4695,
doi:10.1093/nar/gkg694.
6.Vvedenskaya, I. O., Vahedian-Movahed, H., Zhang,
Y., Taylor, D. M., Ebright, R. H., and Nickels, B. E. (2016)
Interactions between RNA polymerase and the core recognition element
are a determinant of transcription start site selection, Proc. Natl.
Acad. Sci. USA, 113, E2899-2905,
doi: 10.1073/pnas.1603271113.
7.Kulbachinskiy, A., and Mustaev, A. (2006) Region
3.2 of the sigma subunit contributes to the binding of the
3′-initiating nucleotide in the RNA polymerase active center and
facilitates promoter clearance during initiation, J. Biol.
Chem., 281, 18273-18276,
doi: 10.1074/jbc.C600060200.
8.Pupov, D., Kuzin, I., Bass, I., and Kulbachinskiy,
A. (2014) Distinct functions of the RNA polymerase sigma subunit region
3.2 in RNA priming and promoter escape, Nucleic Acids Res.,
42, 4494-4504, doi: 10.1093/nar/gkt1384.
9.Duchi, D., Bauer, D. L., Fernandez, L., Evans, G.,
Robb, N., Hwang, L. C., Gryte, K., Tomescu, A., Zawadzki, P.,
Morichaud, Z., Brodolin, K., and Kapanidis, A. N. (2016) RNA polymerase
pausing during initial transcription, Mol. Cell, 63,
939-950, doi: 10.1016/j.molcel.2016.08.011.
10.Dulin, D., Bauer, D. L. V., Malinen, A. M.,
Bakermans, J. J. W., Kaller, M., Morichaud, Z., Petushkov, I., Depken,
M., Brodolin, K., Kulbachinskiy, A., and Kapanidis, A. N. (2018)
Pausing controls branching between productive and non-productive
pathways during initial transcription in bacteria, Nat. Commun.,
9, 1478, doi: 10.1038/s41467-018-03902-9.
11.Vvedenskaya, I. O., Vahedian-Movahed, H., Bird,
J. G., Knoblauch, J. G., Goldman, S. R., Zhang, Y., Ebright, R. H., and
Nickels, B. E. (2014) Transcription. Interactions between RNA
polymerase and the “core recognition element” counteract
pausing, Science, 344, 1285-1289,
doi: 10.1126/science.1253458.
12.Petushkov, I., Pupov, D., Bass, I., and
Kulbachinskiy, A. (2015) Mutations in the CRE pocket of bacterial RNA
polymerase affect multiple steps of transcription, Nucleic Acids
Res., 43, 5798-5809, doi: 10.1093/nar/gkv504.
13.Wiesler, S. C., Weinzierl, R. O., and Buck, M.
(2013) An aromatic residue switch in enhancer-dependent bacterial RNA
polymerase controls transcription intermediate complex activity,
Nucleic Acids Res., 41, 5874-5886,
doi: 10.1093/nar/gkt271.
14.Mekler, V., Kortkhonjia, E., Mukhopadhyay, J.,
Knight, J., Revyakin, A., Kapanidis, A. N., Niu, W., Ebright, Y. W.,
Levy, R., and Ebright, R. H. (2002) Structural organization of
bacterial RNA polymerase holoenzyme and the RNA polymerase-promoter
open complex, Cell, 108, 599-614.
15.Straney, D. C., and Crothers, D. M. (1987) A
stressed intermediate in the formation of stably initiated RNA chains
at the Escherichia coli lacUV5 promoter, J. Mol.
Biol., 193, 267-278,
doi: 10.1016/0022-2836(87)90218-x.
16.Kapanidis, A. N., Margeat, E., Ho, S. O.,
Kortkhonjia, E., Weiss, S., and Ebright, R. H. (2006) Initial
transcription by RNA polymerase proceeds through a DNA-scrunching
mechanism, Science, 314, 1144-1147,
doi: 10.1126/science.1131399.
17.Revyakin, A., Liu, C., Ebright, R. H., and
Strick, T. R. (2006) Abortive initiation and productive initiation by
RNA polymerase involve DNA scrunching, Science, 314,
1139-1143, doi: 10.1126/science.1131398.
18.Henderson, K. L., Felth, L. C., Molzahn, C. M.,
Shkel, I., Wang, S., Chhabra, M., Ruff, E. F., Bieter, L., Kraft, J.
E., and Record, M. T., Jr. (2017) Mechanism of transcription initiation
and promoter escape by E. coli RNA polymerase, Proc. Natl.
Acad. Sci. USA, 114, E3032-E3040,
doi: 10.1073/pnas.1618675114.
19.Winkelman, J. T., and Gourse, R. L. (2017) Open
complex DNA scrunching: a key to transcription start site selection and
promoter escape, BioEssays, 39,
doi: 10.1002/bies.201600193.
20.Carpousis, A. J., and Gralla, J. D. (1985)
Interaction of RNA polymerase with lacUV5 promoter DNA during
mRNA initiation and elongation. Footprinting, methylation, and
rifampicin-sensitivity changes accompanying transcription initiation,
J. Mol. Biol., 183, 165-177,
doi: 10.1016/0022-2836(85)90210-4.
21.Ko, J., and Heyduk, T. (2014) Kinetics of
promoter escape by bacterial RNA polymerase: effects of promoter
contacts and transcription bubble collapse, Biochem. J.,
463, 135-144, doi: 10.1042/BJ20140179.
22.Samanta, S., and Martin, C. T. (2013) Insights
into the mechanism of initial transcription in Escherichia coli
RNA polymerase, J. Biol. Chem., 288, 31993-32003,
doi: 10.1074/jbc.M113.497669.
23.Murakami, K. S., Masuda, S., and Darst, S. A.
(2002) Structural basis of transcription initiation: RNA polymerase
holoenzyme at 4 Å resolution, Science, 296,
1280-1284, doi: 10.1126/science.1069594.
24.Li, L., Molodtsov, V., Lin, W., Ebright, R. H.,
and Zhang, Y. (2020) RNA extension drives a stepwise displacement of an
initiation-factor structural module in initial transcription, Proc.
Natl. Acad. Sci. USA, 117, 5801-5809,
doi: 10.1073/pnas.1920747117.
25.Petushkov, I., Esyunina, D., Mekler, V.,
Severinov, K., Pupov, D., and Kulbachinskiy, A. (2017) Interplay
between sigma region 3.2 and secondary channel factors during promoter
escape by bacterial RNA polymerase, Biochem. J., 474,
4053-4064, doi: 10.1042/BCJ20170436.
26.Kammerer, W., Deuschle, U., Gentz, R., and
Bujard, H. (1986) Functional dissection of Escherichia coli
promoters: information in the transcribed region is involved in late
steps of the overall process, EMBO J., 5, 2995-3000.
27.Hsu, L. M., Cobb, I. M., Ozmore, J. R., Khoo, M.,
Nahm, G., Xia, L., Bao, Y., and Ahn, C. (2006) Initial transcribed
sequence mutations specifically affect promoter escape properties,
Biochemistry, 45, 8841-8854,
doi: 10.1021/bi060247u.
28.Hsu, L. M. (2002) Promoter clearance and escape
in prokaryotes, Biochim. Biophys. Acta, 1577, 191-207,
doi: 10.1016/s0167-4781(02)00452-9.
29.Hsu, L. M. (2009) Monitoring abortive initiation,
Methods, 47, 25-36,
doi: 10.1016/j.ymeth.2008.10.010.
30.Vo, N. V., Hsu, L. M., Kane, C. M., and
Chamberlin, M. J. (2003) In vitro studies of transcript
initiation by Escherichia coli RNA polymerase. 3. Influences of
individual DNA elements within the promoter recognition region on
abortive initiation and promoter escape, Biochemistry,
42, 3798-3811, doi: 10.1021/bi026962v.
31.Heyduk, E., and Heyduk, T. (2018) DNA template
sequence control of bacterial RNA polymerase escape from the promoter,
Nucleic Acids Res., 46, 4469-4486,
doi: 10.1093/nar/gky172.
32.Lerner, E., Chung, S., Allen, B. L., Wang, S.,
Lee, J., Lu, S. W., Grimaud, L. W., Ingargiola, A., Michalet, X.,
Alhadid, Y., Borukhov, S., Strick, T. R., Taatjes, D. J., and Weiss, S.
(2016) Backtracked and paused transcription initiation intermediate of
Escherichia coli RNA polymerase, Proc. Natl. Acad. Sci.
USA, 113, E6562-E6571,
doi: 10.1073/pnas.1605038113.
33.Mekler, V., Pavlova, O., and Severinov, K. (2011)
The interaction of E. coli RNA polymerase σ70
subunit with promoter elements in the context of free
σ70, RNA polymerase holoenzyme and the
β′–σ70 complex, J. Biol.
Chem., 286, 270-279, doi: 10.1074/jbc.M110.174102.
34.Mekler, V., and Severinov, K. (2015) Use of RNA
polymerase molecular beacon assay to measure RNA polymerase
interactions with model promoter fragments, Methods Mol. Biol.,
1276, 199-210, doi: 10.1007/978-1-4939-2392-2_11.
35.Svetlov, V., and Artsimovitch, I. (2015)
Purification of bacterial RNA polymerase: tools and protocols,
Methods Mol. Biol., 1276, 13-29,
doi: 10.1007/978-1-4939-2392-2_2.
36.Petushkov, I., Esyunina, D., and Kulbachinskiy,
A. (2017) σ38-Dependent promoter-proximal pausing by bacterial
RNA polymerase, Nucleic Acids Res., 45, 3006-3016,
doi: 10.1093/nar/gkw1213.
37.Pupov, D., Petushkov, I., Esyunina, D., Murakami,
K. S., and Kulbachinskiy, A. (2018) Region 3.2 of the σ factor
controls the stability of rRNA promoter complexes and potentiates their
repression by DksA, Nucleic Acids Res., 46, 11477-11487,
doi: 10.1093/nar/gky919.
38.Pupov, D., Esyunina, D., Feklistov, A., and
Kulbachinskiy, A. (2013) Single-strand promoter traps for bacterial RNA
polymerase, Biochem. J., 452, 241-248,
doi: 10.1042/BJ20130069.
39.Pupov, D., Miropolskaya, N., Sevostyanova, A.,
Bass, I., Artsimovitch, I., and Kulbachinskiy, A. (2010) Multiple roles
of the RNA polymerase β′ SW2 region in transcription
initiation, promoter escape, and RNA elongation, Nucleic Acids
Res., 38, 5784-5796, doi: 10.1093/nar/gkq355.
40.Mekler, V., Minakhin, L., Kuznedelov, K.,
Mukhamedyarov, D., and Severinov, K. (2012) RNA
polymerase–promoter interactions determining different stability
of the Escherichia coli and Thermus aquaticus
transcription initiation complexes, Nucleic Acids Res.,
40, 11352-11362, doi: 10.1093/nar/gks973.
41.Mekler, V., Minakhin, L., and Severinov, K.
(2011) A critical role of downstream RNA polymerase–promoter
interactions in the formation of initiation complex, J. Biol.
Chem., 286, 22600-22608,
doi: 10.1074/jbc.M111.247080.
42.Sugimoto, N., Nakano, S., Katoh, M., Matsumura,
A., Nakamuta, H., Ohmichi, T., Yoneyama, M., and Sasaki, M. (1995)
Thermodynamic parameters to predict stability of RNA/DNA hybrid
duplexes, Biochemistry, 34, 11211-11216,
doi: 10.1021/bi00035a029.