Mathematical Modeling Reveals the Importance of the DED Filament Composition in the Effects of Small Molecules Targeting Caspase-8/c-FLIPL Heterodimer
N. V. Ivanisenko1,a and I. N. Lavrik1,2,b*
1Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia2Translational Inflammation Research, Medical Faculty, Otto von Guericke University Magdeburg, 39106 Magdeburg, Germany
* To whom correspondence should be addressed.
Received July 17, 2020; Revised July 24, 2020; Accepted July 24, 2020
Procaspase-8 activation at the death-inducing signaling complex (DISC) triggers extrinsic apoptotic pathway. Procaspase-8 activation takes place in the death effector domain (DED) filaments and is regulated by c-FLIP proteins, in particular, by the long isoform c-FLIPL. Recently, the first-in-class chemical probe targeting the caspase-8/c-FLIPL heterodimer was reported. This rationally designed small molecule, FLIPin, enhances caspase-8 activity after initial heterodimer processing. Here, we used a kinetic mathematical model to gain an insight into the mechanisms of FLIPin action in a complex with DISC, in particular, to unravel the effects of FLIPin at different stoichiometry and composition of the DED filament. Analysis of this model has identified the optimal c-FLIPL to procaspase-8 ratios in different cellular landscapes favoring the activity of FLIPin. We predicted that the activity FLIPin is regulated via different mechanisms upon c-FLIPL downregulation or upregulation. Our study demonstrates that a combination of mathematical modeling with system pharmacology allows development of more efficient therapeutic approaches and prediction of optimal treatment strategies.
KEY WORDS: apoptosis, DISC, caspase-8, FLIPin, DED, death receptorsDOI: 10.1134/S0006297920100028
Abbreviations: DED, death effector domain; DISC, death-inducing signaling complex; DR, death receptor; FLIPin, FLIP inhibitor.
INTRODUCTION
The extrinsic apoptotic pathway is initiated by the activation of death receptors (DRs), in particular, CD95/Fas/APO-1, or TRAIL-R1/2 [1]. This leads to the formation of a macromolecular platform termed death-inducing signaling complex (DISC) [2, 3]. The DISC consists of the corresponding DR, FADD (Fas-associated protein with death domain), procaspases-8 and -10, and c-FLIP. DISC formation induces procaspase-8 activation, which takes place at the death effector domain (DED) filaments. The interactions between DEDs of FADD, procaspases-8/10, and c-FLIP contribute to the assembly of DED filaments that serve as a platform for dimerization and subsequent activation of procaspase-8 [4-7].
In the course of dimerization, conformational changes occurring in procaspase-8a/b lead to the rearrangement of the so-called L2 loop [8, 9]. This event allows formation of the active site of caspase-8, thereby initiating procaspase-8 activation. Importantly, activation of procaspase-8 is followed by the auto-catalytical processing which involves the cleavage of the L2 loop at D374 into the L2 (“processed” L2) and L2′ fragments. This leads to the generation of procaspase-8a/b cleavage products p43/p41 and p12 [10]. These products are further auto-catalytically processed via proteolysis at D384 and D210/216 to the active caspase-8 subunits p10 and p18, which form the heterotetramer p102/p182 [11-14]. The procaspase-8a/b homodimer, as well as its cleavage product p43/p41, possess the catalytic activity that is closely linked to the formation of the enzyme active center upon procaspase-8 dimerization [7, 11]. Procaspase-8 substrate specificity differ from the substrate specificity of the caspase-8 heterotetramer p102/p182 [7, 11].
Three c-FLIP isoforms, named long (L), short (S), and Raji (R) (c-FLIPL, c-FLIPS, and c-FLIPR, respectively) have been reported [15, 16]. Short c-FLIP isoforms, c-FLIPS and c-FLIPR, block procaspase-8 activation at the DED filaments, while c-FLIPL at the DISC can act in both pro- and anti-apoptotic manner [8, 9, 17-19]. From the structural point of view, the pro-apoptotic function of c-FLIPL is mediated by the formation of the procaspase-8/c-FLIPL heterodimers in which the L2 loop of procaspase-8 is stabilized in the closed conformation through interactions with c-FLIPL. This leads to an increase in the enzymatic activity of caspase-8 [8, 20, 21]. The pro-apoptotic action of c-FLIPL is observed at low or intermediate levels of protein expression, while at the high expression levels, c-FLIPL is anti-apoptotic [17]. In addition to the studies demonstrating that the level of c-FLIPL expression plays a key role in its pro- versus anti-apoptotic function, there is recent evidence that the anti-apoptotic role of c-FLIPL can be explained by its influence on the composition and structure of the DED filament [18, 22]. In particular, overexpression of c-FLIP terminates the growth of the DED filaments leading to their shortening and restriction of caspase-8 activation. In this way, the quantity of c-FLIP plays a role of a “control checkpoint” of the DED filament length and architecture [18].
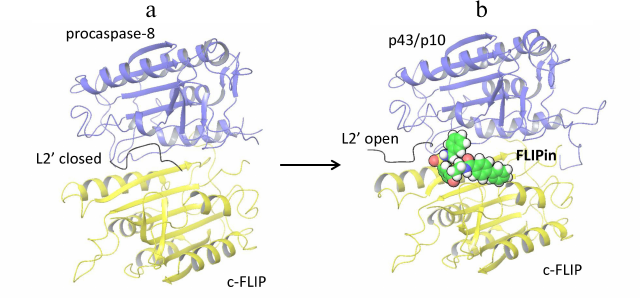
Procaspase-8 activation and its subsequent cleavage at D374 in the procaspase-8/c-FLIPL heterodimer compromises the closed conformation of the procaspase-8 L2 loop, which results in the inhibition of caspase-8 activity in the procaspase-8/c-FLIPL heterodimer [8]. Recently, a small molecule named FLIPin (FLIP inhibitor) was constructed, that was aimed to rescue the loss of the heterodimer catalytic activity [23]. FLIPin was designed to bind to c-FLIPL at the caspase-8/c-FLIPL heterodimer interface and to mimic the stabilizing effect of the L2′ loop in the closed conformation (Fig. 1). In particular, it was hypothesized that FLIPin would bind to the heterodimer after procaspase-8 processing to p43/p41 and thereby restore the interaction network leading to the stabilization of the active center of p43/p41. This compound has been obtained via in silico virtual screening. The optimized compound FLIPinBγ enhanced caspase-8 activity at the DISC and promoted activation of the effector caspases and DR-mediated apoptosis.
Fig. 1. Structural model of caspase-8/c-FLIPL heterodimer bound to FLIPin. a) Structure of procaspase-8 (dark blue) and c-FLIPL (yellow) heterodimer [PDB ID 3H11]. The L2′ loop in the closed conformation is indicated. b) Structural model of of the c-FLIPL [PDB ID 3H11] and processed caspase-8-p43/p10 [PDB ID 3H13] heterodimer bound to FLIPin (green) at the putative binding site. L2′ loop in the open conformation is indicated.
Mathematical modeling of the apoptotic network has become a powerful tool to unravel the dynamics of cell death regulation [24, 25]. In particular, the CD95 signaling network has been studied in detail using ordinary differential equations (ODEs) and other mathematical formalisms such as agent-based models [5, 26-28]. The mathematical models revealed a threshold behavior of CD95 signaling and provided a quantitative insight into the pro-apoptotic role of c-FLIPL, as well as molecular details of the cross-talk between pro- and anti-apoptotic decisions at CD95 [26, 29, 30].
Recently, we have developed a mathematical model to analyze the effects of FLIPin on the procaspase-8 processing at the DISC [23]. However, this model did not take into account the recently reported role of c-FLIP in the regulation of DISC composition and stoichiometry [18]. Here, to account for these new structural insights into the role of c-FLIPL and to gain quantitative understanding of the mechanisms of FLIPin action, we used mathematical modeling with ODEs. This allowed us to predict the optimal ratios between procaspase-8 and c-FLIPL, as well as a cellular landscape favorable for the FLIPin action. Our results provide the basis for the development of new, more efficient therapeutic anti-cancer approaches, as well as allow to further elucidate the mechanisms of DED filament regulation.
MATERIALS AND METHODS
Modeling. The used mathematical model was based on a system of ODEs for caspase-8 activation at the DISC with or without FLIPinBγ described in [23] and further developed in this study. Reaction rates and model parameters were taken from [23]. Scipy.integrate.odeint from Python SciPy package (www.scipy.org) was used to solve the ODEs.
The model was described by 12 differential equations (1-12, Table 1) and 8 parameters (Table 2):
Equations of the model:
(1)
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
Table 1. Designations used in the model and initial values
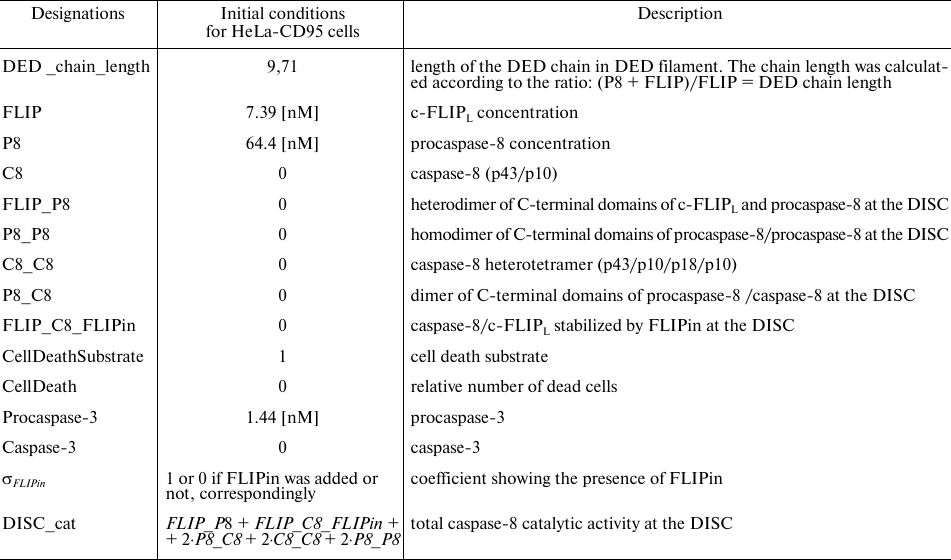
Table 2. Model parameters. Parameter values
were taken from [23]

RESULTS
ODE model of caspase-8 activation at the DED filament accounting for the effects of FLIPin. FLIPin targets the caspase-8/c-FLIPL heterodimer. Hence, the number of caspase-8/c-FLIPL heterodimers formed in the DED filament should directly correlate with the activity of FLIPin. Recently, we have shown that c-FLIPL has two specific binding sites in the DED filament and, therefore, there are two specific sites where the caspase-8/c-FLIPL heterodimer can be assembled [18]. In this regard, c-FLIPL can interact with the FADD DED, as well as with the procaspase-8 DED2. This leads to a scenario in which c-FLIPL can limit the DED filament growth and, in the case of overexpression, shorten DED filaments [18]. Hence, we aimed at developing an ODE model that would analyze the effects of FLIPin considering both factors: the number of caspase-8/c-FLIPL heterodimers and their structural position in the DED filament. In particular, we focused on how FLIPin activity depends on the DED filament length and the c-FLIPL : procaspase-8 ratio.
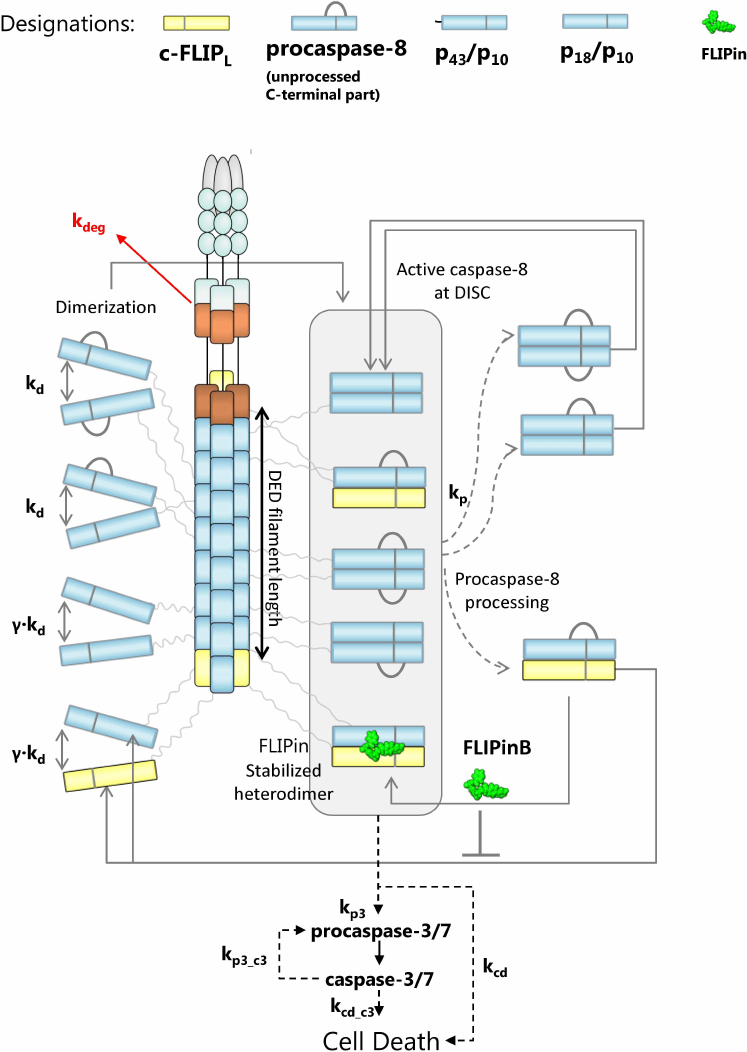
To approach these questions, we selected a recently constructed ODE model of FLIPin action on the caspase-8/c-FLIPL heterodimer and further developed it under consideration of new structural information on the DED filaments. For this purpose, we introduced an additional parameter, which defined the DED chain length. We took into account that a DED filament is composed of three DED chains and, on average, there is one c-FLIPL molecule per DED chain (according to recent proteomics analyses [18]). The topology of the model included formation of the homo- (procaspase-8/procaspase-8) and heterodimers (procaspase-8/c-FLIPL) at the DED filaments (Fig. 2). After homo- and heterodimerization, procaspase-8 undergoes autocatalytic activation in the DED filament, which is followed by the intra- and intermolecular processing of the dimers into p43/p41 (p43-caspase-8), p18, and p10 [10]. For building the model topology, we assumed that the procaspase-8/procaspase-8 homodimer and its cleavage products p43-caspase-8/p43-caspase-8 and p18/p43-caspase-8 have the catalytic activity at the DED filament. These assumptions were based on the earlier reports that the procaspase-8a/b homodimer, as well as its cleavage product p43/p41, possess the catalytic activity that is closely linked to the conformational changes of the active site upon procaspase-8 dimerization and subsequent activation of the dimer [7, 11]. Other assumptions were used for the procaspase-8/c-FLIPL heterodimer. In this case, only the procaspase-8/c-FLIPL heterodimer was considered to have the catalytic activity, while its cleavage product p43-caspase-8/c-FLIPL was assumed to be catalytically inactive and to immediately dissociate from the DED filament in the absence of FLIPin. FLIPin binding was suggested to rescue the p43-caspase-8/c-FLIPL activity to the level of activity typical for the procaspase-8/c-FLIPL heterodimer. Activation of procaspase-8 is accompanied by the caspase-8-mediated cleavage of procaspase-3 to caspase-3 and its subsequent activation, leading to the cell death. In the model, the ‘total cell death level’ corresponded to the level of cleaved cell death substrate, which was integrated over the time. Both caspase-3- and caspase-8-mediated processing of the cell death substrate was incorporated to the model. The binding constants and related parametrization were taken from the previous model of DISC activation upon addition of FLIPin, which was trained on the experimental data from HeLa-CD95 cells [23].
Fig. 2. Mathematical model of caspase-8 activation at the DISC/DED filament considering the action of FLIPin. Model entities and interaction constants are indicated.
In order to model the effects of FLIPin, we utilized the key assumption that c-FLIPL limits the amount of active procaspase-8 in the DISC by terminating the DED filament assembly. At the same time, it was suggested that the increase in the c-FLIPL levels at the DISC does not change the FADD : c-FLIPL ratio. Moreover, it was assumed that DED filaments are assembled directly after CD95L stimulation. The exact stoichiometry of the DED filament was defined by initial conditions of the model. In particular, the ratios between procaspase-8 and c-FLIPL at the DISC were based on the quantitative proteomics analysis performed in our previous work [18]. These studies indicated substoichiometric amounts of c-FLIPL in the DED filament with the c-FLIPL to procaspase-8 ratio of approximately 1 to 9 at the endogenous levels of c-FLIPL expression. However, at a high c-FLIPL expression level, the c-FLIPL to procaspase-8 ratio was approximately 1 to 1 [18]. These ratios were introduced into the model and our next step was to investigate the system behavior in silico.
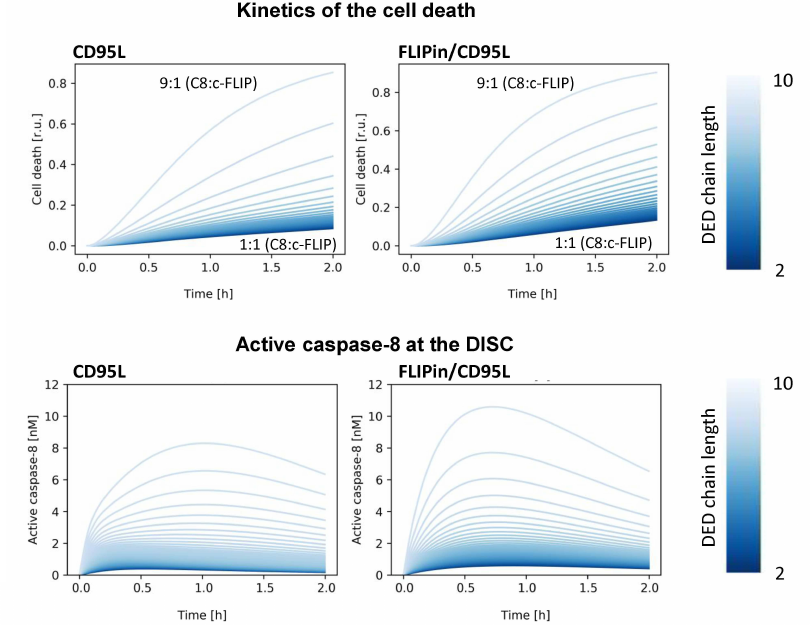
Modeling predicts enhancement of cell death and caspase-8 activity by FLIPin at different c-FLIPL : procaspase-8 ratios. At the first step, we analyzed how the activity of FLIPin depends on different levels of c-FLIPL (Fig. 3). These simulations were carried out considering that the amount of CD95/CD95L/FADD complex does not change with time. First, we investigated the effect of the c-FLIPL level on the catalytic activity of caspase-8 and cell death induction in silico. As expected, the highest caspase-8 activity at the DISC and the highest level of cell death induction were predicted for a low endogenous level of c-FLIPL protein (Fig. 3). In HeLa-CD95 cells, this corresponds to an approximately 1 : 9 c-FLIPL : procaspase-8 ratio at the DISC (Fig. 3). The peak of the caspase-8 activity for these ratios was observed approximately one hour after CD95L stimulation in silico (Fig. 3). An increase in the c-FLIPL level was accompanied by a decrease in the caspase-8 activity at the DISC and suppression of the cell death induction (Fig. 3). At the second step, we investigated the effects of FLIPin. When the cells with the c-FLIPL : procaspase-8 ratio of 1 : 9 were co-treated with FLIPin/CD95L, the peak level of caspase-8 activity was observed at an earlier time point (45 min after CD95L stimulation). Moreover, the total caspase-8 activity after CD95L/FLIPin co-treatment in silico was higher compared to CD95L-only treatment (Fig. 3). Furthermore, promotion of the caspase-8 activity and cell death at the CD95L/FLIPin co-treatment was also observed for all c-FLIPL : procaspase-8 ratios at the DISC (Fig. 3). Therefore, the model has created a profile of sensitivity towards FLIPin that depends on the c-FLIPL levels.
Fig. 3. Kinetics of cell death (top) and caspase-8 activation (bottom) for different caspase-8/c-FLIP ratios. Kinetics for CD95L-only stimulation (left) and CD95L/FLIPin co-stimulation (right) are shown. The color gradient from dark to light indicates the DED chain length from short (1 : 1 c-FLIP : caspase-8 ratio, light color) to long (1 : 9 c-FLIP : caspase-8 ratio, dark color).
Next, we investigated how the length of the DED filaments influences the action of FLIPin. As mentioned above, the key assumption of the model was that an increase in the c-FLIPL level leads to the reduction of the DED filament length. A decrease in the DED filament length results in a decreased amount of procaspase-8 per DED filament. This leads to the reduction in the caspase-8 activity and cell death induction that was observed in silico (Fig. 3). These data fitted well the in vitro observations of Hillert et al. [18]. In particular, a significant decrease in the caspase-8 activity and cell death was observed in HeLa-CD95 cells overexpressing c-FLIPL [18]. Interestingly, under these conditions, the number of the procaspase-8/c-FLIPL heterodimers is naturally expected to increase, but these effects are apparently overridden by the strongly diminished concentration of the procaspase-8 homodimers [18].
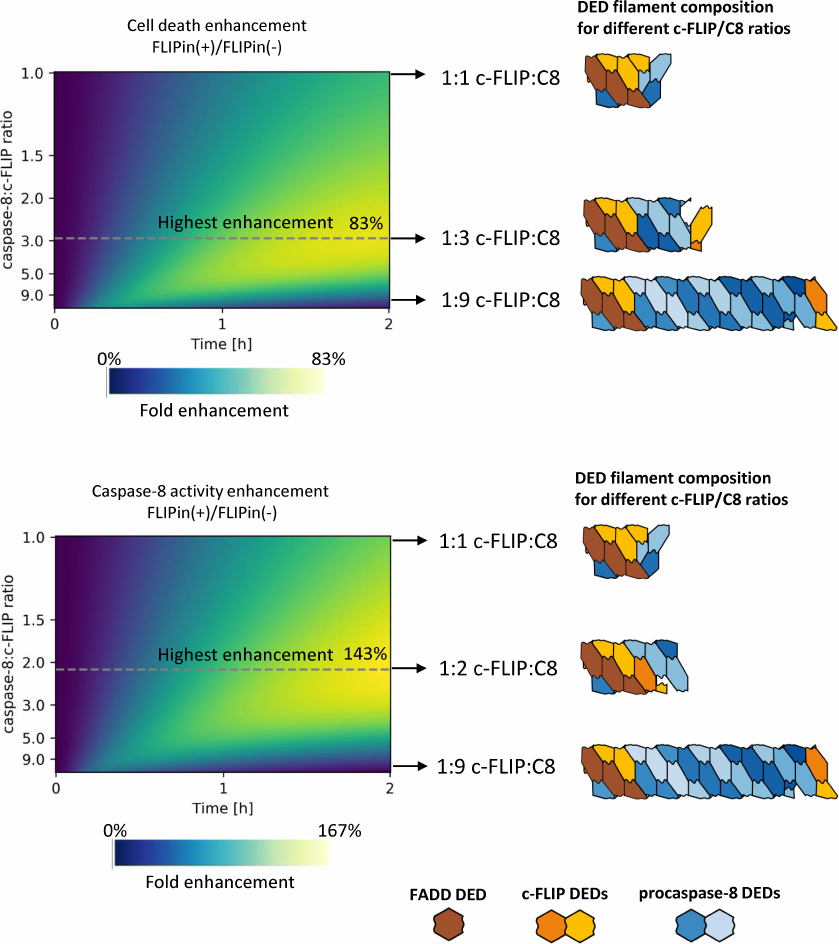
To estimate how FLIPin activity depends on the DED filament length, we introduced a coefficient of FLIPin efficiency. This coefficient was calculated as a ratio between the extent of enhancement of the caspase-8 activity at the DISC and cell death induction upon treatment with CD95L-only and co-treatment with CD95L/FLIPin. The FLIPin efficiency was also estimated at different -FLIPL to procaspase-8 ratios. Then, we calculated the DED filament length and c-FLIPL : procaspase-8 ratio that would correspond to the highest FLIPin efficiency in silico. Strikingly, it was found that the optimal value corresponded to the intermediate level of c-FLIPL expression (Fig. 4). The promotion of the cell death was peaking (>80% increase) at an approximately 1 : 3 c-FLIPL : procaspase-8 ratio, while the highest caspase-8 activity (>140%) was observed at a 1 : 2 c-FLIPL : procaspase-8 ratio (Fig. 4). The model predicted that these c-FLIPL to procaspase-8 ratios provide the optimal ratios between the procaspase-8 homodimers and procaspase-8/c-FLIPL heterodimers resulting in the optimal landscape for the efficient action of FLIPin.
Fig. 4. Relative increase in the extent of cell death (top) and caspase-8 activity (bottom) at different c-FLIP : caspase-8 ratios in the DED filament. The color gradient indicates the fold enhancement by FLIPin. DED filaments corresponding to the highest FLIPin activity are denoted with a dashed line. FADD DEDs, procaspase-8, and c-FLIP are shown in brown, blue, and orange, respectively.
Next, we analyzed how the model predictions correspond to the known cellular systems in which the action of FLIPin has been tested. Based on the results of proteomics analysis, the c-FLIPL : procaspase-8 ratio at the DISC in HeLa-CD95 cells is approximately 1 : 10, resulting in a relatively low level of procaspase-8/c-FLIPL heterodimers [18]. The model predicted that in HeLa-CD95 cells, the contribution of procaspase-8/c-FLIPL heterodimers to the increase in the caspase-8 activity is relatively low. This estimation is valid for all stimulation times, including one hour, the latter corresponding to the predicted maximum of the CD95 DISC activity (Fig. 3). Accordingly, in silico data predicted relatively low efficiency of FLIPin action, which has indeed been observed experimentally [23]. For the DISC of HeLa-CD95 cells, which strongly overexpress c-FLIPL, proteomic analysis showed a 1 : 1 c-FLIP : procaspase-8 ratio [18], resulting in the formation of short DED filaments that likely mostly consist of the procaspase-8/c-FLIPL heterodimers. Interestingly, the predicted peak activity ofcaspase-8 for this cell line corresponds to time intervals that are longer than the estimated half-life of the DISC complex, thus limiting the role of FLIPin in the cell death induction. B lymphoblastoid SKW6.4 cells are another cell line for which a quantitative proteomics analysis of the DISC has been carried out [5, 31, 32]. In this cell line, almost no effect of FLIPin was observed (data not shown), which fits well the stoichiometry of the DISC and DED filaments in SKW6.4 cells. The DED filaments contain very low levels of c-FLIPL, with the c-FLIPL : procaspase-8 ratio similar to that in the DISC of HeLa-CD95 cells (1 : 10). Moreover, SKW6.4 cells are characterized by a faster caspase-8 activation and apoptosis induction compared to HeLa-CD95 cells and with a shorter half-life of the DISC, which apparently precludes detecting the effects of FLIPin on caspase-8 activation. The results of analysis well explain the obtained experimental data and imply further screening of different cell types in order to identify cell lines with the most efficient action of FLIPin.
DISCUSSION
In our previous work, we used advanced methods of in silico computational biology to design a small molecule, FLIPin, targeting the caspase-8/c-FLIPL heterodimer [23]. FLIPin was predicted to mimic the stabilizing effect on the L2′ loop in the closed conformation, leading to an increase in the caspase-8 activity at the DISC and more efficient induction of apoptosis. Experimental data obtained with the optimized small molecule FLIPinBγ support the in silico predicted mechanism of the heterodimer activity [23]. In this study, we used computational modeling to investigate several parameters defining the efficiency of FLIPin action. Our analysis demonstrated that the c-FLIPL to procaspase-8 ratio in the DED filaments is a crucial factor defining this efficiency. This result stems not only from the role of c-FLIPL levels in controlling the ratio of the homo- and heterodimers at the DISC, but is also associated with the essential function of c-FLIPL in regulating the length of the DED filament.
The ratio between the procaspase-8 homo- and heterodimers in the DED filament is controlled by different factors, including expression levels of c-FLIPL and procaspase-8, their association constants at the DISC, and the structure of the DED filament. These factors are linked through a non-linear dynamic resulting in a highly intricate combination that can only be understood at the quantitative level by using computational modeling. The computational model developed in this study allowed us to predict the level of heterodimers depending on the procaspase-8 : c-FLIPL ratio and the DED filament architecture. Moreover, our analysis further confirmed that the level of the heterodimers at the DISC is crucial for the activity of FLIPin. We also showed that FLIPin is controlled by additional factors, such as the architecture of the DED filament. In particular, the model has uncovered the importance of the DED filament architecture for the FLIPin ability to promote cell death and procaspase-8 processing.
Further, we have found that the activity of FLIPin can be strongly influenced by the upregulation of the c-FLIPL protein. However, these effects are non-linear and can only be investigated using modeling. The non-linearity results from the two major effects of c-FLIPL: its effect on the DED filament length and the contribution of procaspase-8/c-FLIPL heterodimers to the total caspase-8 activity of the DISC complex. For example, in HeLa-CD95 cells, there is only a limited number of procaspase-8/c-FLIPL heterodimers compared to caspase-8 homodimers, which limits the effects of FLIPin. Increasing the concentration of c-FLIPL in the DED filament should lead to an increased number of procaspase-8/c-FLIPL heterodimers and increase in the FLIPin activity. In this regard, it was shown by mass-spectrometry analysis that an increase in the c-FLIPL concentrations leads to a higher effective concentration of procaspase-8/c-FLIPL heterodimers [18]. However, this also restricts the DED filament growth, thereby strongly reducing the number of procaspase-8 homodimers and inhibiting the activity of FLIPin. This fits well the experimental observations, as no strong increase in the FLIPin activity was registered in HeLa cells overexpressing c-FLIPL [23].
The optimal c-FLIPL level at the DISC for the FLIPin action was found in silico to be at the c-FLIPL/procaspase-8 ratio between 1 : 9 and 1 : 1. Indeed, in the case of high procaspase-8 content or low c-FLIPL expression, caspase-8 activation occurs quickly, resulting in a very low contribution of the stabilized procaspase-8/c-FLIPL heterodimers to the increase in the procaspase-8 activity. In contrast, in the case of low caspase-8 content or high c-FLIPL expression, the FLIPin activity is limited by short DED filaments, resulting in a reduced caspase-8 activity. Moreover, the current model predicts that the highest activity of FLIPin can be observed when the caspase-8 : c-FLIPL ratio is in a range from 2 : 1 to 3 : 1. The caspase-8 : c-FLIPL ratio of 2 : 1 corresponds to the highest cell death rate, whereas at the 3 : 1 ratio is associated with the highest caspase-8 activity at the DISC. In this regard, we should emphasized that some cancer cells express high levels of c-FLIPL which are close to the quantities predicted by the mathematical model. This suggests that compounds developed based on FLIPin have an extremely high potential as anti-cancer agents [33].
The ODE model has shown that boosting the caspase-8 activity by FLIPin shortly after CD95L stimulation promotes apoptosis induction. The latter might be closely related to a high rate of the DISC complex degradation that was described in [10]. Indeed, the half-life of the DISC in HeLa-CD95 cells was estimated to be only one hour [10], while according to our model, activation of all procaspase-8 molecules at the DISC requires a significantly longer time. In this regard, our approach showed that extending the time of the heterodimer action via the stabilizing effects of FLIPin might serve as a promising strategy for the development of anti-cancer therapies.
Most models of the CD95 DISC that have been constructed so far did not take into account formation of the DED filaments and their architecture [27, 34, 35]. This provides an additional important direction in the future studies on the quantitative dynamics of apoptosis control. Indeed, here we showed an importance of stoichiometry and structure of the DED filament for the efficient apoptosis induction, as well as the necessity for a detailed consideration of the structure of macromolecular complexes of the cell death network in order to understand the life/death decisions in this pathway.
The current model can be considered a minimal model to describe the processing of procaspase-8 at the DISC. The model was trained against experimental data from HeLa cells and fixed CD95L concentration. In the future, a more detailed model can be constructed that will take into account the explicit dynamics of the DED filament formation at different CD95 stimulation strength, as well as the spatial constraints of the c-FLIPL and caspase-8 heterodimerization. Furthermore, from the previous studies, we know that low or so-called threshold concentrations of CD95L regulate the length of the DED chains/filaments [5, 26-28]. A combined influence of both factors on the DED filament architecture and stimulation with the threshold concentrations of CD95L and different c-FLIP levels have to be addressed in the future studies. Moreover, elucidating the intricate interactions of c-FLIPL and procaspase-8 C-terminal domains in the DED filament is an important step towards quantitative understanding of the DED filament dynamics. In particular, interacting DED molecules in the DED filament are likely have a higher efficiency of the C-terminal domain dimerization than the ones located at a distance from each other in the DED filament structure. At the same time, steric constraints likely limit the caspase-8 activity at the short DED filaments that correspond to the 1-2 : 1 : 1 c-FLIP : procaspase-8 : FADD stoichiometry. Explicit modeling of the DED filament assembly and its termination by c-FLIP will reveal a link between the endogenous expression levels of c-FLIP, DISC stoichiometry, and FLIPin activity.
In conclusion, our study shows the importance of combining mathematical modeling with structural modeling and system pharmacology for elucidating molecular mechanisms, as well as developing more efficient therapeutic approaches in personalized medicine.
Funding. This work was supported by the Russian Foundation for Basic Research (projects nos. 19-54-45015, 18-04-00207) and by the Russian State Budget Project (AAAA-A17-117092070032-4).
Ethics declarations. The authors declare that they have no conflicts of interest. This article does not contain any studies involving human participants or animals performed by any of the authors.
REFERENCES
1.Krammer, P. H., Arnold, R., and Lavrik, I. N.
(2007) Life and death in peripheral T cells, Nat. Rev. Immunol.,
7, 532-542.
2.Lavrik, I. N., and Krammer, P. H. (2012) Regulation
of CD95/Fas signaling at the DISC, Cell Death Differ.,
19, 36-41.
3.Zamaraev, A. V., Kopeina, G. S., Zhivotovsky, B.,
and Lavrik, I. N. (2015) Cell death controlling complexes and their
potential therapeutic role, Cell. Mol. Life Sci., 72,
505-517.
4.Dickens, L. S., Boyd, R. S., Jukes-Jones, R.,
Hughes, M. A., Robinson, G. L., et al. (2012) A death effector domain
chain DISC model reveals a crucial role for caspase-8 chain assembly in
mediating apoptotic cell death, Mol. Cell, 47,
291-305.
5.Schleich, K., Warnken, U., Fricker, N., Ozturk, S.,
Richter, P., et al. (2012) Stoichiometry of the CD95 death-inducing
signaling complex: experimental and modeling evidence for a death
effector domain chain model, Mol. Cell, 47, 306-319.
6.Fu, T. M., Li, Y., Lu, A., Li, Z., Vajjhala, P. R.,
et al. (2016) Cryo-EM structure of caspase-8 tandem DED filament
reveals assembly and regulation mechanisms of the death-inducing
signaling complex, Mol. Cell, 64, 236-250.
7.Hughes, M. A., Harper, N., Butterworth, M., Cain,
K., Cohen, G. M., and MacFarlane, M. (2009) Reconstitution of the
death-inducing signaling complex reveals a substrate switch that
determines CD95-mediated death or survival, Mol. Cell,
35, 265-279.
8.Yu, J. W., Jeffrey, P. D., and Shi, Y. (2009)
Mechanism of procaspase-8 activation by c-FLIPL, Proc. Natl. Acad.
Sci. USA, 106, 8169-8174.
9.Micheau, O., Thome, M., Schneider, P., Holler, N.,
Tschopp, J., Nicholson, D. W., Briand, C., and Grutter, M. G. (2002)
The long form of FLIP is an activator of caspase-8 at the Fas
death-inducing signaling complex, J. Biol. Chem., 277,
45162-45171.
10.Kallenberger, S. M., Beaudouin, J., Claus, J.,
Fischer, C., Sorger, P. K., Legewie, S., and Eils, R. (2014) Intra- and
interdimeric caspase-8 self-cleavage controls strength and timing of
CD95-induced apoptosis, Sci. Signal., 7, ra23.
11.Golks, A., Brenner, D., Schmitz, I., Watzl, C.,
Krueger, A., Krammer, P. H., and Lavrik, I. N. (2006) The role of CAP3
in CD95 signaling: new insights into the mechanism of procaspase-8
activation, Cell Death Differ., 13, 489-498.
12.Hoffmann, J. C., Pappa, A., Krammer, P. H., and
Lavrik, I. N. (2009) A new C-terminal cleavage product of procaspase-8,
p30, defines an alternative pathway of procaspase-8 activation, Mol.
Cell Biol., 29, 4431-4440.
13.Lavrik, I., Krueger, A., Schmitz, I., Baumann,
S., Weyd, H., Krammer, P. H., and Kirchhoff, S. (2003) The active
caspase-8 heterotetramer is formed at the CD95 DISC, Cell Death
Differ., 10, 144-145.
14.Ivanisenko, N. V., and Lavrik, I. N. (2019)
Mechanisms of procaspase-8 activation in the extrinsic programmed cell
death pathway, Mol. Biol. (Mosk.), 53, 830-837.
15.Ozturk, S., Schleich, K., and Lavrik, I. N.
(2012) Cellular FLICE-like inhibitory proteins (c-FLIPs): fine-tuners
of life and death decisions, Exp. Cell Res., 318,
1324-1331.
16.Golks, A., Brenner, D., Fritsch, C., Krammer, P.
H., and Lavrik, I. N. (2005) c-FLIPR, a new regulator of death
receptor-induced apoptosis, J. Biol. Chem., 280,
14507-14513.
17.Fricker, N., Beaudouin, J., Richter, P., Eils,
R., Krammer, P. H., and Lavrik, I. N. (2010) Model-based dissection of
CD95 signaling dynamics reveals both a pro- and antiapoptotic role of
c-FLIPL, J. Cell Biol., 190, 377-389.
18.Hillert, L. K., Ivanisenko, N. V., Espe, J.,
Konig, C., Ivanisenko, V. A., Kahne, T., and Lavrik, I. N. (2020) Long
and short isoforms of c-FLIP act as control checkpoints of DED filament
assembly, Oncogene, 39, 1756-1772.
19.Ueffing, N., Keil, E., Freund, C., Kuhne, R.,
Schulze-Osthoff, K., and Schmitz, I. (2008) Mutational analyses of
c-FLIPR, the only murine short FLIP isoform, reveal requirements for
DISC recruitment, Cell Death Differ., 15, 773-782.
20.Boatright, K. M., Deis, C., Denault, J. B.,
Sutherlin, D. P., and Salvesen, G. S. (2004) Activation of caspases-8
and -10 by FLIP(L), Biochem. J., 382, 651-657.
21.Pop, C., Oberst, A., Drag, M., Van Raam, B. J.,
Riedl, S. J., Green, D. R., and Salvesen, G. S. (2011) FLIP(L) induces
caspase 8 activity in the absence of interdomain caspase 8 cleavage and
alters substrate specificity, Biochem. J., 433,
447-457.
22.Hughes, M. A., Powley, I. R., Jukes-Jones, R.,
Horn, S., Feoktistova, M., et al. (2016) Co-operative and hierarchical
binding of c-FLIP and caspase-8: a unified model defines how c-FLIP
isoforms differentially control cell fate, Mol. Cell, 61,
834-849.
23.Hillert, L. K., Ivanisenko, N. V., Busse, D.,
Espe, J., Konig, C., et al. (2020) Dissecting DISC regulation via
pharmacological targeting of caspase-8/c-FLIPL heterodimer, Cell
Death Differ., 27, 2117-2130.
24.Spencer, S. L., and Sorger, P. K. (2011)
Measuring and modeling apoptosis in single cells, Cell,
144, 926-939.
25.Flusberg, D. A., and Sorger, P. K. (2015)
Surviving apoptosis: life-death signaling in single cells, Trends
Cell Biol., 25, 446-458.
26.Bentele, M., Lavrik, I., Ulrich, M., Stosser, S.,
Heermann, D. W., Kalthoff, H., Krammer, P. H., and Eils, R. (2004)
Mathematical modeling reveals threshold mechanism in CD95-induced
apoptosis, J. Cell Biol., 166, 839-851.
27.Lavrik, I. N. (2014) Systems biology of death
receptor networks: live and let die, Cell Death Dis., 5,
e1259.
28.Schleich, K., and Lavrik, I. N. (2013)
Mathematical modeling of apoptosis, Cell Commun. Signal.,
11, 44.
29.Neumann, L., Pforr, C., Beaudouin, J., Pappa, A.,
Fricker, N., Krammer, P. H., Lavrik, I. N., and Eils, R. (2010)
Dynamics within the CD95 death-inducing signaling complex decide life
and death of cells, Mol. Syst. Biol., 6, 352.
30.Buchbinder, J. H., Pischel, D., Sundmacher, K.,
Flassig, R. J., and Lavrik, I. N. (2018) Quantitative single cell
analysis uncovers the life/death decision in CD95 network. PLoS
Comput. Biol., 14, e1006368.
31.Warnken, U., Schleich, K., Schnolzer, M., and
Lavrik, I. (2013) Quantification of high-molecular weight protein
platforms by AQUA mass spectrometry as exemplified for the CD95
Death-Inducing Signaling Complex (DISC), Cells, 2,
476-495.
32.Schleich, K., Buchbinder, J. H., Pietkiewicz, S.,
Kahne, T., Warnken, U., et al. (2016) Molecular architecture of the DED
chains at the DISC: regulation of procaspase-8 activation by short DED
proteins c-FLIP and procaspase-8 prodomain, Cell Death Differ,
23, 681-694.
33.Fulda, S. (2013) Targeting c-FLICE-like
inhibitory protein (CFLAR) in cancer, Expert Opin. Ther.
Targets, 17, 195-201.
34.Spencer, S. L., Gaudet, S., Albeck, J. G., Burke,
J. M., and Sorger, P. K. (2009) Non-genetic origins of cell-to-cell
variability in TRAIL-induced apoptosis, Nature, 459,
428-432.
35.Aldridge, B. B., Gaudet, S., Lauffenburger, D.
A., and Sorger, P. K. (2011) Lyapunov exponents and phase diagrams
reveal multi-factorial control over TRAIL-induced apoptosis, Mol.
Syst. Biol., 7, 553.