The Role of P-Glycoprotein in Decreasing Cell Membranes Permeability during Oxidative Stress
Alexey V. Shchulkin1,a*, Yulia V. Abalenikhina1, Pelageya D. Erokhina1, Ivan V. Chernykh1, and Elena N. Yakusheva1
1Ryazan State Medical University, 390026 Ryazan, Russia* To whom correspondence should be addressed.
Received July 15, 2020; Revised September 3, 2020; Accepted September 3, 2020
P-Glycoprotein (P-gp) is one of the most clinically significant representatives of the ABC transporter superfamily due to its participation in the transport of biotic components and xenobiotics across the plasma membrane. It is known that various chemicals, environmental factors, and pathological processes can affect P-gp activity and expression. In this study, we investigated the role of P-gp in limiting the cell membrane permeability during oxidative stress. Human adenocarcinoma colon cells (Caco-2) overexpressing P-gp were cultured for 72 h in the medium containing hydrogen peroxide (0.1-50 µM). The transport of the P-gp substrate fexofenadine was evaluated in a special Transwell system. The amounts of P-gp and Nrf2 transcription factor were analyzed by the enzyme-linked immunosorbent assay. The concentration of SH-groups in proteins and the contents of lipid peroxidation products and protein carbonyl derivatives were determined spectrophotometrically. Hydrogen peroxide at a concentration of 0.1-5 µM did not significantly affect the studied parameters, while incubation with 10 µM H2O2 decreased in the level of SH groups in cell lysates and increased in the amount of Nrf2 in the cell lysates. Nrf2, in its turn, mediated an increase in the content and activity of the P-gp transporter, thus limiting the increasing permeability of the cell membrane. Hydrogen peroxide at a concentration of 50 µM promoted oxidative stress, which was manifested as a decrease in the content of SH-groups, increase in the concentration of lipid peroxidation products and protein carbonyl derivatives, and decrease in the P-gp level, which led to a significantly increased permeability of the plasma membrane. These results show that the transport and protective roles of P-gp, in particular, reduction of the cell membrane permeability, are affected by the intensity of oxidative stress and can be manifested only if the extent of membrane damage is insignificant.
KEY WORDS: P-glycoprotein (P-gp), Nrf2, oxidative stress, permeability of biological membranes, hydrogen peroxide cytotoxicity, oxidative damage of lipids and proteins, reduced thiolsDOI: 10.1134/S0006297921020085
Abbreviations: Caco-2, human adenocarcinoma colon cells; DTNB, 5,5′-dithiobis(2-nitrobenzoic acid); ELISA, enzyme-linked immunosorbent assay; HPLC, high-performance liquid chromatography; LPO, lipid peroxidation; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Nrf2, nuclear factor erythroid 2-related factor 2; OD, optical density; P-gp, P-glycoprotein; ROS, reactive oxygen species; UV, ultraviolet.
INTRODUCTION
ATP-binding cassette (ABC) transporters are ATP-dependent proteins that transfer various compounds across the plasma membrane in an energy dependent manner. ABC transporters have been found in all three domains of life – Bacteria, Archaea, and Eukarya, as well as in some giant viruses [1]. Human genome contains 49 genes for ABC transporters that comprise seven subfamilies denoted by Latin letters from A to G. Disruption of the functioning of 18 transporters is associated with the development of various diseases (for example, dysfunction of ABCC7 and ABCB11 transporters causes mucoviscidosis and progressive familial intrahepatic cholestasis, respectively); 20 ABC transporters are involved in the drug transport [2]. ABC transporters have a common structural organization: each transporter molecule consists of two transmembrane domains that form the channel and provide substrate recognition and binding, and two nucleotide-binding domains that hydrolyze ATP [3, 4].
P-glycoprotein (also known as the multidrug resistance protein 1, P-gp, ABCB1 protein, MDR1) is the most investigated protein from the ABC transporter superfamily [5, 6]. P-gp is a large transmembrane glycoprotein with the molecular mass of 170 kDa (with the 150-kDa peptide fragment). P-gp exhibits a wide substrate specificity, although its preferable substrates are lipophilic compounds with the molecular mass of 330-4000 Da that belong to antitumor, hypotensive, and antihistamine agents, cardiac glycosides, antiaggregants, anticoagulants, steroid and thyroid hormones, antibiotics, HIV protease inhibitors, immunosuppressants, and others [7].
P-gp was for the first time isolated by R. L. Juliano and V. Ling in 1976 from the plasma membrane of Chinese hamster ovary cells that were chosen based on their resistance to colchicine and cross-resistance to a wide spectrum of amphiphilic compounds [8]. Later, this protein transporter has been identified in colon enterocytes, hepatocytes, renal tubular epithelial cells, and endothelial cells of the blood-tissue barrier. That is why it is generally accepted nowadays that P-gp plays an important role in the development of tumor resistance to chemotherapy [9], as well as in the transport of endogenous and exogenous compounds [10].
Both the activity and the expression level of P-gp could be affected by various chemical compounds, external environmental factors, and pathological processes [11]. In particular, rifampicin and hypoxia activate P-gp, while cyclosporine and verapamil inhibit it [7], which, in turn, affects the penetration of its substrates into the cells through the plasma membrane (enhances or reduces it, respectively).
One of the factors affecting the plasma membrane permeability is the overproduction of reactive oxygen species (ROS). ROS cause activation of lipid peroxidation (LPO) in the membrane, which results in pore formation, reduction of the bilayer thickness, disruption of membrane fluidity, and, finally, increase in the membrane permeability [12].
On the other hand, excessive generation of ROS and oxidative stress development promote expression of the redox-sensitive Nrf2 transcription factor (nuclear factor erythroid 2-related factor 2) [13], which, in turn, could upregulate P-gp expression [14], thereby controlling membrane permeability for the P-gp substrates.
Therefore, the permeability of the plasma membrane for the P-gp substrates could either increase (due to the membrane disruption) or decrease (due to the enhanced P-gp activity). Investigation of this not yet elucidated problem is the subject of this work.
MATERIALS AND METHODS
Culturing of Caco-2 cells. Human colon adenocarcinoma (Caco-2) cells (Collection of Vertebrate Cell Cultures Center for Collective Use, St. Petersburg, Russia) were cultured at 37°C in 5% CO2 in a WS-189C incubator (World Science, South Korea) in DMEM medium with a high glucose content (4500 mg/liter) supplemented with L-glutamine (4 mM), 15% bovine fetal serum, penicillin (100 U/ml), and streptomycin (100 µg/ml) (all components from Sigma-Aldrich, Germany). After reaching 70-90% confluency, the cells were detached from the flask surface by adding trypsin-EDTA solution (0.25% trypsin, 0.2% EDTA; Sigma-Aldrich) and plated into 96-well microplates (Corning, USA) for evaluation of hydrogen peroxide toxicity, 6-well microplates (Corning, USA) for estimation of the hydrogen peroxide effect on the content of P-gp, Nrf2, LPO products, protein carbonyl derivatives, and thiol SH-groups in proteins, or special 12-mm Transwell® plates with polycarbonate membrane inserts with 0.4-µm pores (Corning, USA) for evaluation of the effect of hydrogen peroxide on the cell membrane permeability for the P-gp substrate [15]. The cells were cultured for 21 day, which was the time interval required for their spontaneous differentiation into small intestine enterocyte-like cells overexpressing P-gp. The culture medium was changed every other day, and then every day during the last week of culturing.
Oxidative stress was modeled by adding hydrogen peroxide (Sigma-Aldrich) to the culture medium to the final concentration of 0.1, 0.5, 1, 5, 10, and 50 µM followed by 72-h incubation. The culture medium was changed daily. Each experiment was performed in triplicate.
Survival test (cytotoxicity test). The cells were plated into a 96-well microplate (104 cells per well) and cultured for 21 days; next, the cells were incubated with H2O2 for 72 h. On the completion of incubation, 20 µl of 0.5% 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was added to each well followed by 2-h incubation. The MTT solution was then aspirated and 200 µl of 1% dimethyl sulfoxide solution (PanEko, Russia) [16] was added. Absorption was recorded after 10 min at 530 nm with a Stat Fax 2100 plate reader (Awareness Technology, USA) [16].
Survival of Caco-2 cells in the presence of hydrogen peroxide was calculated according to the equation: (OD of test wells – OD of medium)/(OD of control wells – OD of medium)×100%, where OD is the optical density.
Preparation of cell lysates for analysis. The cells were plated into 6-well microplates (105 cells per well) and cultured for 21 days; H2O2 was then added to the culture medium for 72 h. On the completion of incubation, the cells were detached from the well surface with trypsin–EDTA solution (0.25% trypsin, 0.2% EDTA; Sigma-Aldrich).
A fraction of the cells (2 × 106) was washed three times with phosphate buffer (pH 7.4; PanEko), lysed using triple freeze-thaw cycle in 200 µl of the buffer at –20°C, and used for enzyme-linked immunosorbent assay (ELISA).
The remaining cells (1 × 106) were washed with isotonic NaCl solution (Medpro, Russia) and mixed with 150 µl of lysing buffer [50 mM Tris-HCl, pH 7.4, 150 mM KCl, 0.5% Triton X-100, protease inhibitor mixture (2 mM AEBSF, 0.3 µM aprotinin, 130 µM bestatin, 1 mM EDTA, 14 µM E-64, 1 µM leupeptin)], mixed on a shaker, and incubated on ice for 10 min. Next, the sample was centrifuged for 10 min at 5000g (CM-50, Eppendorf, Germany); the cytoplasmic (extranuclear) fraction was transferred into separate tubes and used for determination of the thiol (SH–) group concentration.
The samples for determining the concentration of LPO products and carbonyl protein derivatives (3 × 106 cells per 450 µl of lysing buffer) were obtained by the same procedure [17].
Analysis of P-gp and Nrf2 content in Caco-2 cells. The content of P-gp and Nrf2 in the cell lysates was determined using commercial ELISA kits (Human permeability glycoprotein ELISA Kit and Human nuclear factor erythroid 2-related factor 2 ELISA Kit, respectively; Blue gene, China). Absorption was recorded at 450 nm with a Stat Fax 2100 plate reader (Awareness Technology). The total protein amount in the samples was determined with the Bradford assay (Pierce Coomassie Plus Assay Kit, ThermoFisher, USA).
The concentration of thiol (SH-) groups in proteins was determined from the difference between the total amount of thiol groups and number of SH-groups associated with low-molecular-weight compounds. The total amount of thiol groups in lysates was determined using the Ellman method with 5,5′-dithiobis(2-nitro)-benzoic acid (DTNB) under non-denaturing conditions [18]. DTNB (2 mM, 100 µl; Serva, Germany) in 1 M Tris-HCl buffer (pH 8.0) and 1 ml of distilled water were added to 100-µl lysate sample. Following 30-min incubation, the amount of formed 5-thio-2-nitrobenzoic acid was evaluated from the absorption at 412 nm with a Stat Fax 2100 plate reader (Awareness Technology). The concentration of SH-groups was calculated based on the extinction coefficient ε412 = 13.6 mM–1·cm–1 [19]. In order to determine the content of SH-groups in low-molecular-weight compounds, the sample was first mixed with cold 5% trichloroacetic acid (Chimmed, Russia), incubated on ice for 15 min, and centrifuged at 11,000g (CM-50, Eppendorf) for 5 min at 4°C. The supernatant was neutralized with 1% NaOH (Chimmed) and used for determination of SH-groups in low-molecular-weight compounds from the reaction with DTNB.
The concentration of LPO products and protein carbonyl derivatives was determined with the commercial kits from Elabscience (China) and Sigma-Aldrich, respectively.
The method for identifying the LPO products (malondialdehyde and 4-hydroxyolefins 4-hydroxy-2-nonenal and 4-hydroxy-2-hexanal) is based on their reaction with 1-methyl-2-phenylindole resulting in the formation of a stable chromophore with the absorption maximum at 586 nm [20].
The method for the evaluation of protein carbonyl derivatives is based on their reaction with 2,4-dinitrophenylhydrazine producing 2,4-dinitrophenylhydrazones, which are detected from the absorption at 375 nm. The concentration of protein carbonyl derivatives was calculated using the extinction coefficient ε375 = 22 mM–1·cm–1 [21].
Analysis was carried out with a Stat Fax 2100 plate reader (Awareness Technology).
Evaluation of the P-gp substrate (fexofenadine) transport across the plasma membrane of Caco-2 cells. The penetration of the P-gp substrate fexofenadine through the bilayer lipid membrane in Caco-2 cells was evaluated using special Transwell inserts (Fig. 1). A Transwell insert separates two compartments in a well: apical compartment and basolateral compartment. The Caco-2 cells were seeded onto the bottom of the apical compartment made of a semipermeable membrane at a density of 105 cells/cm2 and cultured for 21 days.
Fig. 1. Schematic representation of a Transwell insert. The insert separates the apical and the basolateral chambers. The bottom of the apical chamber is made of a semipermeable membrane onto which Caco-2 cells were seeded at a density of 105 cells/cm2.
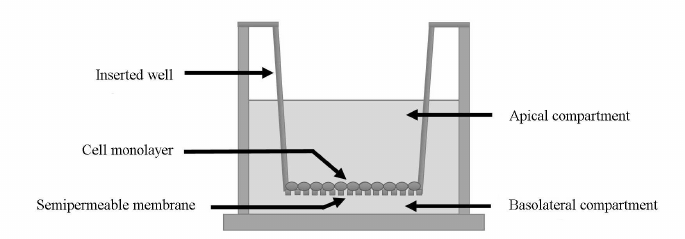
The integrity of the cell monolayer was evaluated from the transepithelial resistance, which was determined with a Millicell ERS-2 voltohmmeter (Millipore, USA). The experiments on the substrate transport by P-gp were carried out when the transepithelial resistance reached above 500 mOhm·cm2. The culture medium with H2O2 at the tested concentration was added to the wells. On the completion of incubation, the culture medium was replaced with the transport medium (Hanks solution, Sigma-Aldrich) containing 25 mM HEPES (Sigma-Aldrich) and 1% dimethyl sulfoxide (PanEko). Next, fexofenadine (Sigma-Aldrich) was added to the apical chamber to the final concentration of 150 µM [22]. The samples were taken from the basolateral recipient chamber 1, 2, and 3 h after substrate addition to the apical chamber and assayed for the fexofenadine concentration (a-b transport due to the diffusion against the P-gp-mediated transport). The transport of fexofenadine from the basolateral chamber to the apical chamber (b-a transport due to the diffusion and P-gp functioning) was assessed under similar condition. For this purpose, the substrate was added to the basolateral chamber at the same concentration, and the samples were taken from the apical chamber 1, 2, and 3 h after the substrate addition to determine fexofenadine concentration.
The substrate transport was evaluated using the following equation [23]:
Papp = (dQ/dt) × (1/A × CO),
where Papp is the apparent permeability coefficient (cm/s); dQ/dt is the change in the substrate amount in the recipient chamber during the incubation time (µM·ml/s); A is the surface area of the semipermeable membrane in the Transwell insert (cm2); C0 is the initial substrate concentration in the donor chamber (µM).
Fexofenadine concentration in the transport medium was determined by high-performance liquid chromatography (HPLC) on a Synergi 4 µm Polar-RP 80 Å column (250×4.6; Phenomenex, USA) with UV detection at 220 nm using a Staier chromatograph (Russia) according to the original procedure [24]. A sample of the transport medium (50 µl) containing fexofenadine was diluted in 150 µl of mobile phase and 100 µl of the obtained solution was loaded into the chromatograph. The separation temperature was 45°C; the flow rate was 1 ml/min. The mobile phase contained 128 ml of acetonitrile (PanReac AppliChem, Spain), 267.4 ml of deionized water, and 6.33 ml of glacial acetic acid (PanReac AppliChem); triethylamine was added to pH 6.7. The retention time for fexofenadine under these conditions was 12.8 min. The amount of fexofenadine was determined using the absolute calibration technique from the areas under the peaks. The analytical range of this technique was 1.2-57.4 µM.
Statistical analysis. The obtained results were analyzed with the StatSoft Statistica 13.0 software (USA, license no. JPZ811I521319AR25ACD-W) and Microsoft Excel for Mac ver. 16.24 (ID02984-001-000001). The results are presented as mean ± standard deviation (M ± SD). To estimate the significance of differences, dispersion analysis (ANOVA) was used; the pairwise comparison was performed using the Newman–Keuls procedure. The differences were considered significant at p-value less than 0.05. The correlation analysis was performed based on the Pearson coefficient.
RESULTS
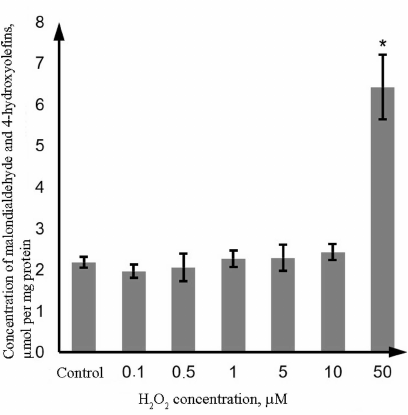
Oxidative damage of proteins and lipids in the Caco-2 cells exposed to H2O2. Oxidative damage of lipids was evaluated based on the level of LPO products (malondialdehyde and 4-hydroxyolefins); oxidative damage of proteins was evaluated from the content of thiol (SH-) groups in proteins and concentration of protein carbonyl derivatives. The obtained results show that H2O2 at the concentration of 0.1-10 µM did not affect the level of LPO products; however, in the presence of 50 µM H2O2, this parameter increased by 194.9% (p = 0.0002) in comparison with the control (Fig. 2).
Fig. 2. Changes in the concentration of LPO products (malondialdehyde and 4-hydroxyolefins) in the lysate of Caco-2 cells during oxidative stress induced by incubation with 0.1-50 µM hydrogen peroxide for 72 h as determined by the photometric technique. The data are shown as M ± SD (n = 3); (*) significant difference from the control, p < 0.05.
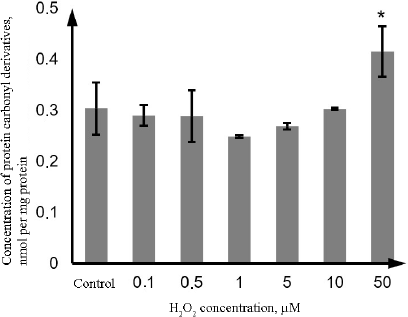
Similarly, the content of the protein carbonyl derivatives in the lysate of Caco-2 cells increased only at the H2O2 concentration of 50 µM (by 36.5%, p = 0.001) (Fig. 3).
Fig. 3. Changes in the concentration of protein carbonyl derivatives during oxidative stress induced by incubation with 0.1-50 µM hydrogen peroxide for 72 h as determined by the photometric technique. The data are shown as M ± SD (n = 3); (*) significant difference from the control, p < 0.05.
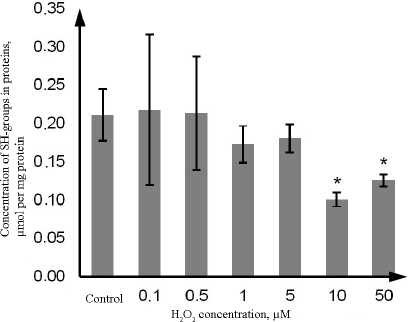
At the same time, the content of SH-groups in proteins decreased by 52.4% (p = 0.021) upon exposure to 10 µM H2O2, and by 38.1% (p = 0.022) upon exposure to 50 µM H2O2 in comparison with the control. Hydrogen peroxide at the concentration of 0.1, 0.5, 1, and 5 µM did not affect the level of reduced thiol groups vs. the control (Fig. 4)
Fig. 4. Changes in the concentration of thiol groups in proteins during oxidative stress induced by incubation with 0.1-50 µM hydrogen peroxide for 72 h as determined by the photometric technique using rection with DTNB. The data are shown as M ± SD (n = 3); (*) significant difference from the control; p < 0.05.
Hence, H2O2 at the concentration of 50 µM caused lipid and protein damage in Caco-2 cells; moreover, oxidation of SH-groups (which might play a regulatory role) occurred already at the H2O2 concentration of 10 µM.
Cytotoxic effect of H2O2 on Caco-2 cells was evaluated using the MTT assay. The survival of intact cells (control group) was 100%, while the survival rate of cells exposed to 0.1-10 µM hydrogen peroxide was 87.6 ± 9.3%, which was not significantly different from the control group. Following exposure to 50 µM H2O2, the survival rate decreased to 58.8 ± 11.5% (p = 0.02). Taken together with the results described above, these data indicate that H2O2 causes damage and death of cells only in the case of severe oxidative stress manifested as lipid and protein damage.
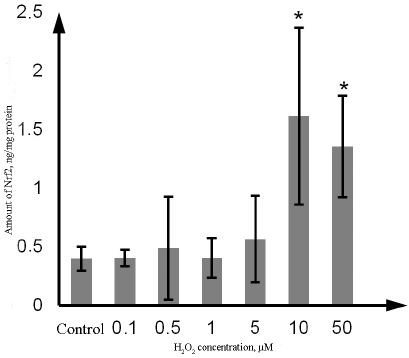
Effect of H2O2 on P-gp and Nrf2 content. The amount of Nrf2 in the Caco-2 cells incubated with H2O2 at the concentration of 10 µM and 50 µM increased by 302.5% (p = 0.02) and 237.5% (p = 0.03), respectively, in comparison with the control. No significant changes were observed at lower H2O2 concentrations (Fig. 5).
Fig. 5. Changes in the Nrf2 content in the lysate of Caco-2 cells during oxidative stress induced by exposure to 0.1-50 µM hydrogen peroxide for 72 h as determined by ELISA. The data are shown as M ± SD (n = 3); (*) significant difference from the control; p < 0.05.
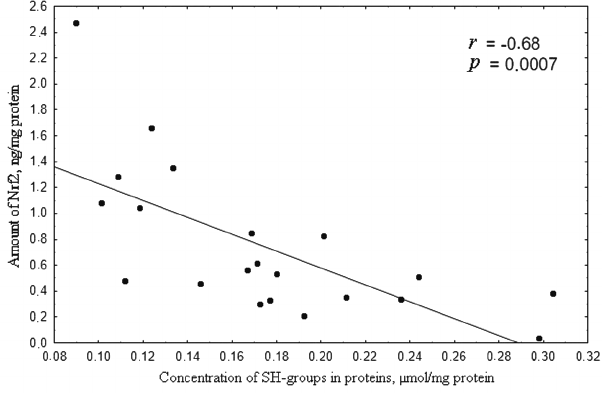
In the 0.1 to 50 µM range of H2O2 concentration, the level of Nrf2 was inversely proportional to the content of thiol groups in proteins (Fig. 6), which is in agreement with the classic notion on the mechanism of Nrf2 regulation [25].
Fig. 6. Correlation between the concentration of SH-groups (µmol/mg protein) and amount of Nrf2 (ng/mg protein) upon induction of oxidative stress by 0.1-50 µM hydrogen peroxide for 72 h. Correlation coefficient r = –0.68; p = 0.0007, which indicates inverse dependence of the concentration of reduced thiols on the amount of Nrf2.
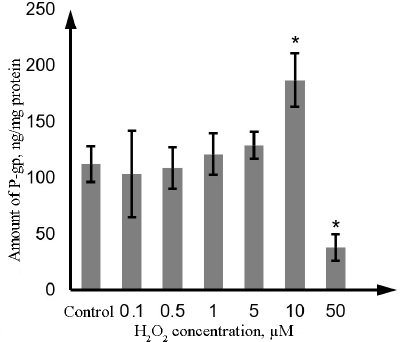
As estimated by ELISA, the amount of P-gp in Caco-2 cells was 110.8 ng/mg protein or 23.5 ng/106 cells. H2O2 at the concentration of 0.1, 0.5, 1, and 5 µM did not affect the amount of P-gp. Incubation with 10 µM hydrogen peroxide increased the P-gp content by 68.9% (p = 0.01), whereas exposure to 50 µM hydrogen peroxide decreased it by 65.8% (p = 0.01) in comparison with the control samples (Fig. 7).
Fig. 7. Changes in the P-gp content in the lysate of Caco-2 cells during oxidative stress induced by exposure to 0.1-50 µM hydrogen peroxide for 72 h as determined by ELISA. The data are shown as M ± SD (n = 3); (*) significant difference from the control; p < 0.05.
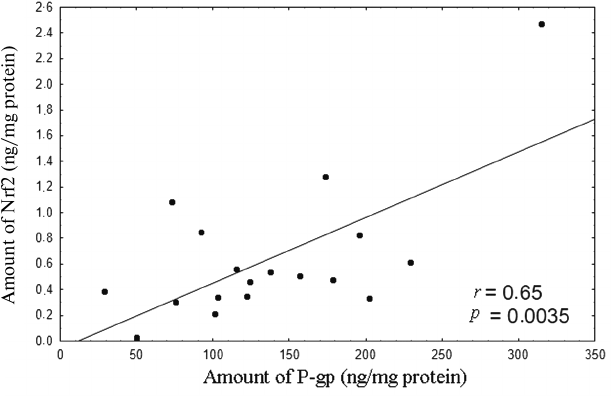
Analysis of the results revealed that within the 0.1 to 10 µM H2O2 concentration range, the level of P-gp in the lysates of Caco-2 cells was directly proportional to the Nrf2 content (Fig. 8).
Fig. 8. Correlation between the amount of Nrf2 (ng/mg of protein) and amount of P-gp (ng/mg of protein) upon induction of oxidative stress by 0.1-10 µM hydrogen peroxide for 72 h. The correlation coefficient r = 0.65, p = 0.0035, indicates direct dependence between the amounts of Nrf2 and P-gp under conditions ensuring plasma membrane integrity.
Hence, oxidative stress and oxidation of SH-groups increase the level of the Nrf2 transcription factor, which, in turn, increases the amount of the protein transporter P-gp. Further development of oxidative stress leads to the decrease in the amount of P-gp, while the level of Nrf2 remains upregulated.
Effect of H2O2 on the transport of the P-gp substrate fexofenadine through the bilayer lipid membrane in Caco-2 cells. The determined apparent permeability coefficient Papp b-a for fexofenadine characterizing its transport from the basolateral chamber into the apical chamber due to the diffusion and P-gp activity was 3.08 · 10–6 ± 0.94 · 10–6 cm/s, and the apparent permeability coefficient Papp a-b describing the transport from the apical chamber to the basolateral chamber due to the diffusion against the P-gp-mediated transport was 1.13 · 10–6 ± 0.6 · 10–6 cm/s. It can be seen from the presented data that the transport due to the P-gp activity was almost three times faster than the transport due to the diffusion against the P-gp-mediated transport (p = 0.013) (table).
Effect of H2O2 on the transport of P-gp substrate
(fexofenadine) through the bilayer lipid membrane of Caco-2 cells
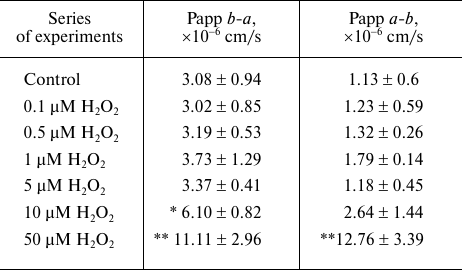
Notes. Following exposure to hydrogen peroxide (72 h), Caco-2 cells were
incubated in the transport medium with fexofenadine (150 µM) in
the apical or basolateral chamber of the corresponding Transwell
inserts. The aliquots were taken after 1, 2, and 3 h of incubation from
the recipient basolateral chamber (a-b transport) or
apical chamber (b-a transport) and assayed for the
fexofenadine concentration by HPLC with UV detection; (*) difference
from the control, p = 0.056, M ± SD, n = 3; (**)
significant difference from the control, p < 0.01.
H2O2 at the concentrations of 0.1-5 µM did not affect the transport of fexofenadine from the apical to the basolateral chamber and in the opposite direction. At the same time, an advantageously higher rate of transport due to the diffusion and P-gp activity vs. the transport rate due to the diffusion against the P-gp-mediated transport was maintained (p < 0.05), which implied the stability of the P-gp activity and membrane permeability.
H2O2 at a concentration of 10 µM caused an increase in the apparent permeability coefficient Papp b-a for fexofenadine by 98.1% (p = 0.056) in comparison with the control but did not affect the apparent permeability coefficient Papp a-b, suggesting that fexofenadine transport by P-gp increased, while diffusion against the operating protein transporter did not change significantly.
The increase in the H2O2 concentration to 50 µM resulted in the increase in the apparent permeability coefficients Papp b-a and Papp a-b by 260.7% (p = 0.001) and 1029.2% (p = 0.001), respectively, in comparison with the control (table). In the process, Papp b-a (due to P-gp activity and diffusion) became equal to Papp a-b (due to diffusion against protein transporter activity) (p > 0.05).
Hence, it was shown that 10 µM H2O2 increased the activity of P-gp in Caco-2 cells. The damage of both cell membrane and P-gp occurred at higher H2O2 concentrations, resulting in a significant increase in the cell membrane permeability in both directions.
DISCUSSION
P-gp is an ATP-dependent protein transporter responsible for the efflux of exogenous and endogenous compounds from the cell to the extracellular medium and biological fluids. In tumor cells, it provides the defense against cytostatics; in intestine – prevents absorption of biotic compounds and xenobiotics; in endothelium of the blood-brain barrier – ensures permeation of substrates into the brain tissue; in liver and kidneys – facilitates release of substances into the bile and the urine [6]. Some therapeutic preparations and environmental factors could change the biosynthesis rate and the activity of P-gp. In particular, it was shown that Nrf2 stimulates P-gp synthesis during oxidative damage in hepatocytes [26], neurons [27], cells of the blood-brain barrier [28], and lung carcinoma A549 cells [14], which results in the upregulation of the P-gp activity and, consequently, decrease in the membrane permeability for its substrates.
In this study, the amount of P-gp and the permeability of the bilayer lipid membrane for its substrate fexofenadine during oxidative stress of varying degree caused by H2O2 were evaluated in the in vitro experiments in the Caco-2 cell line. The lifetime of hydrogen peroxide in cells is milliseconds to seconds, which allows H2O2 to penetrate into the cell and to exert its intracellular effects. The penetration of hydrogen peroxide through the cell membrane could be mediated by aquaporins [29]. H2O2 is a stable compound with a low reactivity; that is why it can function as a secondary messenger [30]. Uncontrolled interaction of hydrogen peroxide with transition metals (Fe2+ or Cu+) in the Fenton reaction results in the ROS formation [31]. ROS can be neutralized by the antioxidant systems or accumulated in the cells causing oxidative stress, which leads to protein, lipid, and DNA damage, mutagenesis, and cell death. The most sensitive to oxidative damage are sulfur-containing amino acid residues (methionine and cysteine), which are also important for the regulatory functions of proteins [32, 33].
The change in the ratio of reduced and oxidized SH-groups in proteins affects the redox-sensitive transcription factor Nrf2. Its expression is upregulated during oxidative stress in order to ensure cell defense against the effects of free radicals [13, 34]. Under normal conditions, this transcription factor exists in a complex with the repressor protein Keap1; their association is regulated by a number of protein kinases. On one hand, Keap1 facilitates ubiquitination and proteasomal degradation of Nrf2 (which requires the presence of two cysteine residues in the Keap1 molecule); on the other hand, Keap1 prevents Nrf2 transport form the cytoplasm to the nucleus [35]. The Keap1-Nrf2 complex dissociates following activation, and Nrf2 is translocated to the nucleus, where it binds to the antioxidant response elements (AREs) and activates transcription of defense enzymes [35].
At the same time, oxidative stress leads to the increased permeability of the bilayer lipid membrane [36]. The nature and the extent of structural and functional damage of the membrane under oxidative stress of varying intensity have not been investigated in sufficient details. To our best knowledge, no studies on the quantitative and functional parameters of the membrane transporter P-gp during oxidative stress development have been conducted so far.
In this work, we demonstrated that the development of oxidative stress caused by 10 µM H2O2 resulted in the oxidation of SH-groups, which upregulated the amount of Nrf2. This transcription factor stimulated increase in the the amount and activity of P-gp, which was manifested as the facilitated transport of the transporter substrate from the basolateral chamber to the apical chamber (due to diffusion and functioning of P-gp) in the used Transwell system. It would be reasonable to expect that in this case, the transport of fexofenadine from the apical chamber to the basolateral chamber would decrease (due to diffusion against the P-gp-mediated transport). However, in our study, this parameter did not change. It is likely that 10 µM H2O2 increased the permeability of the lipid bilayer membrane increases, but the increased activity of P-gp compensated this disturbance.
The increase in the H2O2 concentration to 50 µM further promoted the oxidative stress and caused accumulation of malonic dialdehyde and 4-hydroxyolefins (4-hydroxy-2-nonenal and 4-hydroxy-2-haxanal) and protein carbonyl derivatives, as well as reduced the amount of P-gp. The level of Nrf2 remained increased, which was most likely due to the localization of the investigated proteins. P-gp is located predominantly in the plasma membrane and is readily accessible to exogenously added H2O2, while Nrf2 is located in the cytoplasm and the nucleus [37] and, therefore, is more protected from the effects of H2O2 in the incubation medium.
The decrease in the amount of P-gp and the damage of the lipid bilayer membrane under oxidative stress condition (50 µM H2O2) activated fexofenadine transport across the plasma membrane from the basolateral chamber to the apical chamber and in the opposite direction. Furthermore, the value of the apparent permeability coefficient Papp b-a became almost the same as the value of the apparent permeability coefficient Papp a-b. The biochemical changes occurring upon exposure to 50 µM H2O2 were accompanied by a significant increase in the cell death rate (according to the results of the MTT assay).
The obtained results could be of great practical importance. For example, it is commonly accepted that P-gp is one of the key factors limiting the penetration drugs into the brain through the blood-brain barrier and therefore, reducing the efficiency of treatment of brain disorders (acute disruption of cerebral circulation, epilepsy). Hence, P-gp inhibition is considered as a possible therapeutic approach in the treatment of these pathologies [38, 39].
It was shown in our study that the oxidative stress development (which is commonly observed in neurological diseases) resulted in the damage of lipid bilayer membrane and P-gp, which was accompanied by the increase in the plasma membrane permeability. Therefore, the protective role of P-gp in pathology is maintained only in the case of compensated oxidative stress, which puts in doubt the eligibility of P-gp as a promising target for therapeutic intervention.
The increase in the amount of Nrf2 during oxidative stress upregulates the amount of P-gp, which limits the increasing permeability of the cell membrane. Further oxidative stress development causes damage to the membrane and P-gp, resulting in a significant increase in the plasma membrane permeability in both directions. This implies that the transport and protective roles of P-gp, in particular, reduction of the cell membrane permeability, depend on the degree of oxidative stress and are manifested only in the case of insignificant membrane damage.
Funding. This work was supported by the Grant from the President of the Russian Federation for Support of Young Scientists (project no. MK-1856.2020.7).
Ethics declarations. The authors declare no conflict of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
REFERENCES
1.Davidson, A. L. (2008) Structure, function, and
evolution of bacterial ATP-binding cassette systems, Microbiol. Mol.
Biol. Rev., 72, 317-364, doi: 10.1128/MMBR.00031-07.
2.Vasiliou, V., Vasiliou, K., and Nebert, D. W.
(2009) Human ATP-binding cassette (ABC) transporter family, Hum.
Genomics, 3, 281-290, doi: 10.1186/1479-7364-3-3-281.
3.Licht, A., and Schneider, E. (2011) ATP binding
cassette systems: Structures, mechanisms, and functions, Cent. Eur.
J. Biol., 6, 785-801, doi: 10.2478/s11535-011-0054-4.
4.Esser, L., Zhou, F., Pluchino, K. M., Shiloach, J.,
Ma, J., et al. (2017) Structures of the multidrug transporter
P-glycoprotein reveal asymmetric ATP binding and the mechanism of
polyspecificity, J. Biol. Chem., 292, 446-461, doi:
10.1074/jbc.M116.755884.
5.Sharom, F. J. (2011) The P-glycoprotein multidrug
transporter, Essays Biochem., 50, 161-178, doi:
10.1042/bse0500161.
6.Yakusheva, E. N., and Titov, D. S. (2018) Structure
and function of multidrug resistance protein 1, Biochemistry
(Moscow), 83, 907-929, doi: 10.1134/S0006297918080047.
7.Kukes, V. G., Grachev, S. V., Sychev, D. A., and
Ramenskaya, G. V. (2008) Metabolism of Drugs. Scientific Basis of
Personalized Medicine: Guide for Doctors, Geotar-Media, Moscow.
8.Juliano, R. L., and Ling, V. (1976) A surface
glycoprotein modulating drug permeability in Chinese hamster ovary cell
mutants, Biochim. Biophis. Acta, 455, 155-162, doi:
10.1016/0005-2736(76)90160-7.
9.Pokharel, D., Roseblade, A., Oenarto, V., Lu, J.
F., and Bebawy, M. (2017) Proteins regulating the intercellular
transfer and function of P-glycoprotein in multidrug-resistant cancer,
Ecancermedicalscience, 11, e768, doi:
10.3332/ecancer.2017.768.
10.Borst, P., and Schinkel, A. H. (2013)
P-glycoprotein ABCB1: a major player in drug handling by mammals, J.
Clin. Invest., 23, 4131-4133, doi: 10.1172/JCI70430.
11.Yano, K., Tomono, T., and Ogihara, T. (2018)
Advances in studies of P-glycoprotein and its expression regulators,
Biol. Pharm. Bull., 41, 11-19, doi:
10.1248/bpb.b17-00725.
12.Van der Paal, J., Neyts, E. C., Verlackt, C. C.
W., and Bogaerts, A. (2016) Effect of lipid peroxidation on membrane
permeability of cancer and normal cells subjected to oxidative stress,
Chem. Sci., 7, 489-498, doi: 10.1039/C5SC02311D.
13.Raghunath, A., Sundarraj, K., Nagarajan, R.,
Arfuso, F., Bian, J., et al. (2018) Antioxidant response elements:
discovery, classes, regulation and potential applications, Redox.
Biol., 17, 297-314, doi: 10.1016/j.redox.2018.05.002.
14.Wu, B., Li, H. X., Lian, J., Guo, Y. J., Tang, Y.
H., et al. (2019) Nrf2 overexpression protects against paraquat induced
A549 cell injury primarily by upregulating P glycoprotein and reducing
intracellular paraquat accumulation, Exp. Ther. Med., 17,
1240-1247, doi: 10.3892/etm.2018.7044.
15.Yakusheva, E. N., Shchulkin, A. V., Chrnykh, I.
V., Popova, N. M., Kotlyarova, A. A., and Slepnev, A. A. (2019)
Assessment of drugs belonging to inhibitors and inductors of
p-glycoprotein in vitro, Obz. Klin. Biokhim. Lekkarst.
Ter., 17, 71-78, doi: 10.7816/RCF17171-78.
16.Tolosa, L., Donato, M. T., and
Gómez-Lechón, M. J. (2015) General cytotoxicity
assessment by means of the MTT assay, Methods Mol. Biol.,
1250, 333-348, doi: 10.1007/978-1-4939-2074-7_26.
17.Tinnikov, A. A., and Samuels, H. H. (2013) A
novel cell lysis approach reveals that caspase-2 rapidly translocates
from the nucleus to the cytoplasm in response to apoptotic stimuli,
PLoS One, 8, e61085, doi:
10.1371/journal.pone.0061085.
18.Boschi-Muller, S., Azza, S., Sanglier-Cianferani,
S., Talfournier, F., Dorsselear, A. V., and Branlant, G. (2000) A
sulfenic acid enzyme intermediate is involved in the catalytic
mechanism of peptide methionine sulfoxide reductase from
Escherichia coli, J. Biol. Chem., 275,
35908-35913, doi: 10.1074/jbc.M006137200.
19.Ellman, L. G. (1959) Tissue sulfhydryl groups,
Arch. Biochem. Biophys., 82, 70-77, doi:
10.1016/0003-9861(59)90090-6.
20.Gérard-Monnier, D., Erdelmeier, I.,
Régnard, K., Moze-Henry, N., Yadan, J. C., Chaudière, J.
(1998) Reactions of 1-methyl-2-phenylindole with malondialdehyde and
4-hydroxyalkenals. Analytical applications to a colorimetric assay of
lipid peroxidation, Chem. Res. Toxicol., 11, 1176-1183,
doi: 10.1021/tx9701790.
21.Weber, D., Davies, M. J., and Grunea, T. (2015)
Determination of protein carbonyls in plasma, cell extracts, tissue
homogenates, isolated proteins: Focus on sample preparation and
derivatization conditions, Redox Biol., 5, 367-380, doi:
10.1016/j.redox.2015.06.005.
22.Petri, N., Tannergren, C., Rungstad, D., and
Lennernäs, H. (2004) Transport Characteristics of Fexofenadine in
the Caco-2 Cell Model, Pharmac. Res., 21, 1398-1404, doi:
10.1023/B:PHAM.0000036913.90332.b1.
23.Elsby, R., Surry, D. D., Smith, V. N., and Gray,
A. J. (2008) Validation and application of Caco-2 assays for the in
vitro evaluation of development candidate drugs as substrates or
inhibitors of P-glycoprotein to support regulatory submissions,
Xenobiotica, 38, 1140-1164, doi:
10.1080/00498250802050880.
24.Erokhina, P. D., Abalenikhina, Yu. V., Shchulkin,
A. V., Chrnykh, I. V., Popova, N. M., et al. (2020) Investigation of
the effect of progesterone on activity of P-glycoprotein in
vitro, I. P. Pavlov Rus. Med. Biol. Herald, 28,
135-142, doi: 10.23888/PAVLOVJ2020282135-142.
25.Lennicke, C., Rahn, J., Lichtenfels, R.,
Wessjohann, L. A., and Seliger, B. (2015) Hydrogen peroxide –
production, fate and role in redox signaling of tumor cells, Cell
Commun. Signal., 13, e39, doi:
10.1186/s12964-015-0118-6.
26.Thakkar, N., Slizgi, J. R., and Brouwer, K. L. R.
(2017) Effect of liver disease on hepatic transporter expression and
function, J. Pharm Sci., 106, 2282-2294, doi:
10.1016/j.xphs.2017.04.053.
27.Grewal, G. K., Kukal, S., Kanojia, N., Saso, L.,
Kukreti, Sh., and Kukreti, R. (2017) Effect of oxidative stress on ABC
transporters: contribution to epilepsy pharmacoresistance,
Molecules, 22, e365, doi: 10.3390/molecules22030365.
28.Aryal, M., Fischer, K., Gentile, C., Gitto, S.,
Zhang, Y.-Z., and McDannold, N. (2017) Effects on P-glycoprotein
expression after blood-brain barrier disruption using focused
ultrasound and microbubbles, PLoS One, 3, e0166061, doi:
10.1371/journal.pone.0166061.
29.Rodrigues, O., Reshetnyak, G., Grondin, A.,
Saijo, Y., Leonhardt, N., et al. (2017) Aquaporins facilitate hydrogen
peroxide entry into guard cells to mediate ABA- and pathogen-triggered
stomatal closure, Proc. Natl. Acad. Sci. USA, 114,
9200-9205, doi: 10.1073/pnas.1704754114.
30.Sies, H. (2017) Hydrogen peroxide as a central
redox signaling molecule in physiological oxidative stress: oxidative
eustress, Redox Biol., 11, 613-619, doi:
10.1016/j.redox.2016.12.035.
31.Smirnoff, N., and Arnaud, D. (2019) Hydrogen
peroxide metabolism and functions in plants, New Phytol.,
2, 1197-1214, doi: 10.1111/nph.15488.
32.Vogelsang, L., and Dietz, K.-J. (2020) Regulatory
thiol oxidation in chloroplast metabolism, oxidative stress response
and environmental signaling in plants, Biochem. J., 477,
1865-1878, doi: 10.1042/BCJ20190124.
33.Poole, L. B. (2015) The basics of thiols and
cysteines in redox biology and chemistry, Free Radic. Biol.
Med., 1, 148-157, doi:
10.1016/j.freeradbiomed.2014.11.013.
34.Kang, K. A., and Hyun, J. W. (2017) Oxidative
stress, Nrf2, and epigenetic modification contribute to anticancer drug
resistance, Toxicol. Res., 33, 1-5, doi:
10.5487/TR.2017.33.1.001.
35.Wen, Zh., Liu, W., Li, X., Chen, W., Liu, J., et
al. (2019) A protective role of the NRF2-Keap1 pathway in maintaining
intestinal barrier function, Oxid. Med. Cell Longev.,
2019, e1759149, doi: 10.1155/2019/1759149.
36.Itri, R., Junqueira, H. C., Mertins, O., and
Baptista, M. S. (2014) Membrane changes under oxidative stress: the
impact of oxidized lipids, Biophys. Rev., 6, 47-61, doi:
10.1007/s12551-013-0128-9.
37.Yoon, D. S., Choi, Y., and Lee, J. W. (2016)
Cellular localization of NRF2 determines the self-renewal and
osteogenic differentiation potential of human MSCs via the
P53–SIRT1 axis, Cell Death Dis., 7, e2093, doi:
10.1038/cddis.2016.3.
38.Fromm, M. F. (2004) Importance of P-glycoprotein
at blood-tissue barriers, Trends Pharmacol. Sci., 25,
423-429, doi: 10.1016/j.tips.2004.06.002.
39.Wang, G.-X., Wang, D.-W., Liu, Y., and Ma, Y.-H.
(2016) Intractable epilepsy and the P-glycoprotein hypothesis, Int.
J. Neurosci., 126, 385-392, doi:
10.3109/00207454.2015.1038710.