CRABP1 and CRABP2 Protein Levels Correlate with Each Other but Do Not Correlate with Sensitivity of Breast Cancer Cells to Retinoic Acid
Adel D. Enikeev1, Andrey V. Komelkov1,a*, Maria E. Axelrod1, Sergey A. Galetsky1, Sergey A. Kuzmichev1, and Elena M. Tchevkina1
1Blokhin National Medical Research Center of Oncology, 115478 Moscow, Russia* To whom correspondence should be addressed.
Received August 17, 2020; Revised September 30, 2020; Accepted September 30, 2020
Retinoic acid (RA) binding proteins, CRABP1 and CRABP2, are molecular chaperones that mediate intracellular activity of RA, the key promoter of cell differentiation with tumor suppressor activity. One of the main functions of CRABP2 is delivery and transfer of RA to the nuclear receptors RAR/RXR, which leads to activation of the transcription of a wide range of retinoid-responsive genes. The functions of CRABP1 are less studied but are apparently associated with sequestration of RA in cytoplasm and limitation of its transcriptional activity, suggesting involvement of this protein in the development of RA resistance. The mechanisms regulating activity of CRABP1 are also poorly understood. Comparison of the CRABP1 level in tumor cell lines of various origins, performed for the first time here, showed absence of the CRABP1 protein in the cell lines of tumors considered to be RA-resistant, and pronounced production of this protein in the RA-sensitive cells. However, analysis carried out with a panel of breast cancer cell lines with different levels of RA-sensitivity showed that there was no correlation between the production of CRABP1 protein and the sensitivity of the cells to RA. At the same time, we found strong correlation between the expression of CRABP1 and CRABP2 proteins in all studied cell types, regardless of their origin and RA-sensitivity/resistance. Moreover, suppression of the CRABP1 level in both RA-sensitive and RA-resistant cells was shown in the cells with cells with knockdown of CRABP2 gene. The revealed CRABP2-dependent regulation of CRABP1 production is a new mechanism of the intracellular retinoic signaling system.
KEY WORDS: retinoic acid, retinoic acid binding proteins, ATRA, CRABP1, CRABP2, proliferation, expression regulationDOI: 10.1134/S0006297921020103
Abbreviations: ATRA, all-trans retinoic acid; BC, breast cancer; CRABP1 and CRABP2, cellular retinoic acid binding proteins 1 and 2; FABP5, fatty acid binding protein 5; GFP, green fluorescent protein; NSCLC, non-small cell lung cancer; PPAR, peroxisome proliferator-activated receptors; RA, retinoic acid; RAR, retinoic acid receptor.
INTRODUCTION
Retinoic acid represents one of the most active intracellular retinol (vitamin A) metabolites. Functional activity of retinoic acid (RA) is associated with transcription regulation of more than five hundred genes containing retinoid responsive elements in the relevant promoter region, which is executed with the help of nuclear RA receptors primarily proteins RAR (RARα, β, and γ) and RXR acting as heterodimeric transcription factors as well as via peroxisome proliferator-activated receptors β/δ (PPARβ/δ) also forming dimer with RXR [1]. RA regulates crucial biological processes related to cell differentiation, tissue remodeling, and immune response through regulation of the transcriptional activity of a number of genes. In the majority of cells, RA stimulates differentiation or apoptosis and could even act as a negative regulator of cell proliferation thereby allowing to consider it as a tumor suppressor. In connection with this, extensive attempts have been made to develop chemotherapeutic agents based on RA and its synthetic analogs. However, clinical use of retinoids is limited only to the treatment of acute promyelocytic leukemia due to rapidly developing resistance of malignant cells to RA [2].
Functional activity of RA is mainly determined by its delivery to the nuclear receptors. Representatives of the intracellular lipid binding protein (iLBP) family, primarily two RA-binding proteins CRABP1 and CRABP2 (cellular retinoic acid binding proteins-1 and -2) as well as representative of the fatty acid-binding protein family (FABP5) are responsible for intracellular transport of the hydrophobic RA molecules to the receptors [3]. According to some data in general the CRABP proteins (or at least CRABP2) ensure interaction of RA with the RAR/RXR receptors, whereas FABP5 delivers RA to the receptor PPARβ/δ. It is assumed that interaction of RA with various nuclear receptors result in the opposite intracellular effects: the CRABP2-dependent RAR/RXR activation stimulated differentiation and the RA-dependent pro-apoptotic and anti-proliferative activity, whereas the FABP5-mediated RA interaction with PPARβ/δ facilitates transcriptional activation of the genes responsible for survival, proliferation, and angiogenesis [4]. RA affinity to CRABPs is higher than to FABP5 [5]. Moreover, in the majority of cells the CRABP2/FABP5 and RAR/PPARβ/δ ratios are shifted towards the CRABP-RAR pathway determining the RA-associated tumor suppressor function in the most of tissue types [6]. In addition, some data exist indicating that the elevated FABP5/CRABP2 ratio correlates with the resistance of cell to RA [7, 8]. In connection with the aforementioned CRABP2 function, this protein is considered as a tumor suppressor. Moreover, CRABP2 has been also shown to exert cytosolic activity in the presence of RA unrelated to activation of its cognate receptors, which also facilitates cell cycle arrest and stabilization of the pro-apoptotic gene transcripts [9, 10]. Despite this, CRABP2 seems to play a tumor-promoting role in some types of tumors [11, 12] via mechanisms remaining poorly understood. The breast cancer (BC) study data demonstrating that FABP5 is mainly expressed in the ER-negative and triple-negative tumors usually displaying RA-resistance [8] indirectly suggest that FABP5 is involved in this phenomenon. CRABP1 demonstrates the highest affinity to RA among the RA-binding proteins [13], but its functional significance has not been elucidated yet, and the available data on its role in cell transformation and tumor progression are contradictory.
It is assumed that binding of CRABP1 to RA results in its cytosolic retention [14], limited activity (including RA access to CRABP2 via a competitive mechanism [15]), and could even contribute to metabolism of the latter [16-18]. In accordance with this, it can be suggested that CRABP1 is involved in acquiring of the cell resistance to RA. However, to our best knowledge there are almost no studies examining a link between CRABP1 and sensitivity to RA except a single work cited above that reported upregulated expression of this protein in the ER/PR-negative breast cancer cells [15]. According to other data CRABP1, similarly to CRABP2, was able to direct RA to RAR receptors, but did not interact with them directly and dissociation of the RA-CRABP1 complex occurred prior to the transfer of RA to the receptor [13]. In this connection, it remains unclear what exactly this protein does in the nucleus, although it carries a nuclear localization signal similar to CRABP2, and its presence in the nucleus has been confirmed in our previous study [19] and reported by others [20].
At present, nothing is known about the potential interplay between the CRABP proteins, which, however, seems quite plausible considering that both proteins facilitate realization of the similar RA-driven events, and are themselves products of the retinoid-responsive genes. Such possibility was suggested previously based on the data obtained in our earlier study [21].
Nonetheless, virtually no studies exist among the numerous publications examining RA-binding proteins that analyze CRABP1 and CRABP2 simultaneously except the aforementioned studies on examining CRABPs in the breast cancer cells [15] and our study investigating non-small cell lung cancer samples [21].
In this work for the first time, we assessed expression of both CRABP1 and CRABP2 proteins in a broad range of cell lines and analyzed its potential link with RA sensitivity. We found lack of stringent association between the production of such proteins and RA sensitivity, albeit the majority of RA-sensitive cell lines was characterized by high expression level, whereas most of the RA-resistant cell lines lacked production of CRABP proteins. In the process, we found correlation between the production of CRABP1 and CRABP2 – all cell lines expressing CRABP1 typically produced high levels of CRABP2, whereas those characterized with the significantly lower or lost expression of CRABP2 did not produce CRABP1. Moreover, for the first time we were able to demonstrate that expression of CRABP proteins is correlated with CRABP2 acting as upstream regulator of CRABP1.
MATERIALS AND METHODS
Cell lines. The following cell lines from ATCC collection were used in the study: non-small cell lung cancer (NSCLC) – A549, H1299, H460; glioblastoma – LN229 and U87; ovarian cancer – OVCAR-8, SK-OV-3, EFO-21; neuroblastoma – SK-N-AS, SH-SY-5Y, IMR-32; BC – MCF7, T47D, SKBR3, HCC1954, MDA-MB-453, HCC1937, MDA-MB-468, MDA-MB-231, HBL100. Pseudoretroviral particles were obtained by using epithelial cell lines 293FT (Invitrogen, USA) and GP293 (Clonetech, USA). Cells were cultured in the DMEM medium (PanEco, Russia) containing 0.294 mg/ml L-glutamine, supplemented with 10% fetal bovine serum (PAA Laboratories, USA), 0.1 mg/ml streptomycin, 100 U/ml penicillin, at 37°C and 5% CO2. MCF10A breast epithelial cells were cultured in the DMEM/F-12 medium (Gibco, USA) supplemented with 5% horse serum (Gibco), 20 ng/ml growth factor EGF (Thermo Fisher Scientific, USA), 0.5 mg/ml hydrocortisone (Sigma-Aldrich, USA), 100 ng/ml cholera toxin (Sigma-Aldrich), and 10 µg/ml insulin (Sigma-Aldrich). Mycoplasma contamination was assessed by using routine PCR protocol followed by staining with Hoechst 33342 (Thermo Fisher Scientific).
Retinoic acid-cell sensitivity. All-trans retinoic acid (ATRA) was dissolved in DMSO for preparing 10 µM stock solution. Cells were seeded 50-300,000 per well depending on the cell line followed by 5-day culturing in the presence of 0.1-100 µM ATRA. After that cells were detached by incubating with 0.25% trypsin-EDTA solution, mixed with a trypan blue dye (1 : 1 cell/dye volume ratio) and counted in a hemocytometer. Cells of the same origin cultured in the presence of a concentration-matched DMSO without ATRA for 5 days were used as controls. Number of live cells in control samples for each cell line was taken as 100%.
Generation of exogenous CRABP1-overexpressing cell lines. Molecular cloning and retrovirus infection were performed according to the previous protocol [22]. Primers specific to the motifs flanking the CRABP1-coding sequence were used for PCR amplification: CRABP1 cloning F: 5′ATTCTCGAGCCACCATGCCCAACTTC3′ and CRABP1 cloning R: 5′ACAGGATCCCTGCCTTCACTCTCGG3′ (restriction sites highlighted in italics, complimentary mRNA sequences underlined). cDNA isolated from the human lung cancer samples and synthesized on the relevant mRNA was used as a PCR template. Amplification was carried out by using high-fidelity DNA Polymerase PFX (Invitrogen), according to the manufacturer’s protocol. CRABP1-coding sequence was cloned into a retrovirus vector pLXSN (Clontech, USA) at restriction sites XhoI and BamHI to be further assessed by using restriction enzymes as well as PCR primers: pLXSN F: CCCTTGAACCTCCTCGTTCG and pLXSN R: TTTCCACACCTGGTTGCTGA, designed complementary to the motifs flanking the insert. Human CRABP1 specificity was verified by sequencing. Pseudoretroviral particles were obtained after GP293 cell transfection performed by using 2 µg DNA equimolar mix pLXSN-CRABP1 and pVSV-G (Clontech) added with LipofectAMINE 2000™ Reagent (Invitrogen), according to the manufacturer’s protocol. After the cells reached 20-30% confluence, 24-, 48- and 72-h virus inoculates were added mixed with the culture medium at 1 : 1 ratio as well as 8 µg/ml polybrene (Sigma-Aldrich). Next, transfected cells were selected by incubating with 1000 µg/ml G418 (Sigma-Aldrich), for 8-9 days. In control, all such cell lines were transfected with the pLXSN-free vector.
Generation of CRABP2 knockdown cell lines. Short hairpin RNA (shRNA) precursors containing CRABP2 sequence were cloned into the lentivirus vector pLKO.1-puro (Addgene, USA) at restriction sites AgeI and EcoRI. The sequences were as follows: sh1CRABP2 – CCGGGAAATGGGAGAGTGAGAATAACTCGAGTTATTCTCACTCTCCCATTTCTTTTTG (TRCN0000021371), sh2CRABP2 – CCGGCGAGGAATTGCTCAAAGTGCTCTCGAGAGCACTTTGAGCAATTCCTCGTTTTTG (TRCN0000021370) (sense and antisense sequences underlined). Final constructs were assessed by using PCR with primers pLKO. 1-seq standard followed by sequencing (ID sequence AH002814.2, Homo sapiens retinoic acid-binding protein II (CRABP-II) gene, complete cds, is available at https://www.ncbi.nlm.nih.gov/nucleotide/AH002814.2).
293FT cells at 70% confluency were used for obtaining pseudoretroviral particles. For this, 2 µg of DNA mix of vectors pLKO. 1 puro, pVSVG (Clontech) and pCMV delta R8.2 (Addgene) at equimolar ratio was used. Transfection was carried out with with Lipofectamine 2000™ Reagent (Invitrogen) according to the manufacturer’s protocol. After reaching 20-30% cell confluency, 24-, 48-, and 72-h virus inoculates were added mixed with culture medium at 1 : 1 ratio and supplemented with 8 µg/ml polybrene (Sigma-Aldrich). Puromycin (Sigma-Aldrich) was used for selection at a dose of 1-2 µg/ml depending on the cell line for 4-5 days. Green fluorescent protein-bearing shRNA (shGFP) was used to transfect parental cell lines in the control group.
Immunoblotting. Immunoblotting was carried out according to the previously published protocol [22]. Cells were lysed by using RIPA buffer (50 mM TRIS-HCl (pH 7.5), 150 mM NaCl, 0.5% DoX, 1% NP-40, 0.1% SDS, 2 mM EDTA) supplemented with a protease inhibitor cocktail (Complete Protease Inhibitor Cocktail, Roche, USA). Protein concentration was measured by using Bradford Protein Assay Kit (Bio-Rad, USA). Next, 5-µg protein sample was separated using 15% PAGE followed by transfer onto a PVDF membrane (Millipore, USA) that was incubated in a 5% BSA blocking solution (PAA Laboratories GmbH) and TBS buffer supplemented with 0.1% TWEEN-20. After that, the membrane was incubated with primary anti-CRABP1 (Sigma-Aldrich, HPA17203; 1 : 1000) or anti-CRABP2 (Sigma-Aldrich, HPA004135; 1 : 500) antibodies for 12 h at 4°C. In control, protein samples were incubated with an anti-β-actin (Abcam, USA, ab8227; 1 : 5000) antibody. In the next step, membranes were washed and incubated with the secondary horseradish peroxidase-conjugated antibodies (Cell Signaling, USA, 29902; 1 : 65,000) for 1 h, at room temperature followed by subsequent washing and processing with an ECL reagent (Enhanced Chemiluminescence; Millipore). Chemiluminescent reaction was monitored using a Kodak GelLogic 2200 Imaging system and analyzed with a Carestream Molecular Imaging Software SE ver. 5.0.1.27.
Statistical analysis. All data were obtained by conducting three independent experiments. The data were presented as mean ± standard deviation (SD), which were processed and plotted by using GraphPad Prizm 8.3 software (GraphPad Software, USA).
RESULTS
CRABP1 and CRABP2 production correlates in cell lines of diverse origins. It was established in our previous work on RA-binding proteins that the levels of CRABP (especially of CRABP1) varied significantly in the cancer cell lines of various origins. Taking into consideration available information on the potential role of CRABP1 as an intracellular RA bioavailability regulator, we suggested that it might be related to the development of RA-resistance. In connection with this, we compared CRABP production in various cancer cell types. In particular, we observed that CRABP1 protein is absent in all cell lines derived from NSCLC (A549, H1299, H460), ovarian cancer (SK-OV-3, OVCAR-8, EFO-21), and glioblastoma (LN229, U87), whereas high levels of this proteins were detected in the neuroblastoma cells (SK-N-AS, SH-SY-5Y, IMR-32) (Fig. 1). Due to the fact that in some cell lines no CRABP1 production was found, we used MCF7 BC cells as a positive control. In addition, we also showed that regardless of the origin all CRABP1-expressing cells also had high levels of CRABP2 protein, whereas the cell types lacking CRABP1 demonstrated noticeably lower or absent CRABP2 expression. Specificity of the antibody-mediated CRABP detection was confirmed by presenting the data after identifying CRABP1 and CRABP2 proteins in the parental cell line H460 lacking endogenous CRABP1 expression and derivative subline showing expression of the exogenous CRABP1 (H460 CRABP1) as well as control cells transfected with the vector lacking specific construct (H460 pLXSN) (Fig. 1). In particular, it was demonstrated that all derivative cell did not produce CRABP2 regardless of the CRABP1 expression, thereby confirming specificity of the antibody-based detection.
Fig. 1. Comparison of production of CRABP1 and CRABP2 in the cancer cell lines of diverse origin using immunoblotting: NSCLC (H1299, A549, H460), ovarian cancer (OVCAR-8, SC-OV-3, EFO-21), glioblastoma (LN229, U87), neuroblastoma (IMR-32, SK-N-AS, SH-SY-5Y). BC cell line MCF7 was used as a positive control for CRABP1 expression. Specificity of anti-CRABP1 and anti-CRABP2 protein antibodies was assessed in the H460 subline derivative overexpressing CRABP1 (H460 CRABP1) or expressing empty vector (H460 pLXSN). Neuroblastoma cell lines are shown twice in the bottom – with (left)/without MCF7 cells (right) to avoid masking differences in the CRABP protein expression between the cell lines at the same membrane exposure time in the presence of the strong signal observed for MCF7 cells.
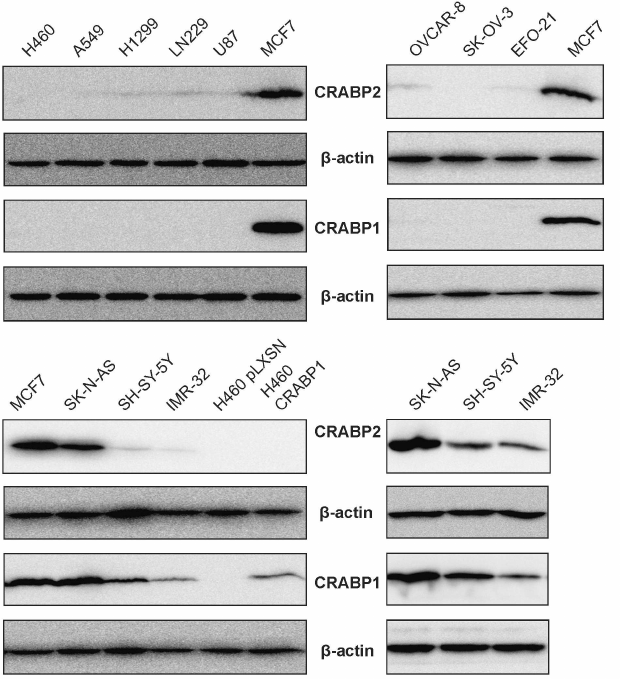
It has been established based on the available information that NSCLC, ovarian cancer, and glioblastoma are considered as RA-resistant cell lines, while neuroblastoma as a RA-sensitive. Thus, we confirmed our assumption that sensitivity of the cells to RA is associated with the level of CRABP1 expression. From the obtained data we further suggest that some regulatory feedback loop might exist between the CRABP1 and CRABP2 proteins. However, both hypotheses should be tested preferably by using cell lines of the same origin that differ in sensitivity to RA.
Comparing retinoic acid-sensitivity of various breast cancer cell lines. An experimental model of breast cancer was chosen to test the aforementioned hypotheses. On the one hand, it was due to availability of a large amount of preclinical data indicating great potential of using RA as a preventive or therapeutic agent for treating this type of tumors [23-25].
On the other hand, BC represents a heterogeneous tumor type both in terms of origin and molecular features (primarily in estrogen and progesterone receptor expression as well as HER2/neu profile), which, according to the literature data also exhibit varying sensitivity to RA [26, 27]. Data on the level of sensitivity of different BC cell lines to RA also vary. It must be mentioned that in different studies different approaches and methods were used to evaluate sensitivity of the cells to RA as well as different RA concentrations. Hence, in the first step it was necessary to compare RA-sensitivity of a broad panel of the BC cell lines under the same conditions in the same experimental series, and select conditions and threshold RA level separating RA-sensitive from RA-resistant cells. We included 9 BC cell lines as well as MCF10A cells derived from the breast epithelial cells usually used as apparently normal (non-tumor) cell line. ATRA (All-Trans-Retinoic Acid, most common and biologically active RA isoform) sensitivity was analyzed by assessing cell proliferation dynamics. For this, each cell line was cultured for 5 days in the standard culture medium (control) or the medium supplemented with ATRA at varying concentrations followed by counting the number of live cells. A range of ATRA concentrations from 0.01 to 100 µM was used. ATRA titration data and analysis of the live cell count allowed to select ATRA threshold concentrations and criteria distinguishing RA-sensitive and RA-resistant cells. Cell lines were considered as RA-sensitive if they demonstrated at least 2-fold decline in cell proliferation after incubation with 10 µM ATRA. This group consisted of the cell lines MCF7, T47D, SKBR3, and HCC1954 (Fig. 2a). Sensitivity of the cell lines to RA within the group also varied: the cell lines SKBR3 and T47D were most sensitive, which showed 2-fold proliferation decline compared to the control even when ATRA was used at 0.1 µM concentration. At the same time, dynamics of cell proliferation in the group consisting of RA-resistant cell lines (MDA-MB-453, MDA-MB-468, MDA-MB-231, HCC1937, HBL100, and MCF10A) did not change after exposure to 10 µM ATRA (Fig. 2b). Moreover, number of live cells in this group did not decrease after incubation with 20 µM ATRA. Nonetheless, it was possible to distinguish cells lines in the group of RA-resistant cell lines demonstrating no change in dynamics of cell proliferation even after being incubated with 50 µM ATRA (HBL100, MDA-MB-231) versus those demonstrating reduced cell proliferation at this ATRA concentration (MDA-MB-453, MDA-MB-468, HCC1937, MCF10A) compared to the control.
Fig. 2. Dynamics of the BC cell line proliferation in the presence of ATRA. Cells were exposed to ATRA at different concentrations for five days followed by calculating percentage of cell survival compared to control (100% cell survival = number of live cells in each cell line cultured for similar time period in the DMEM cell medium supplemented with DMSO at concentrations matching those used to dissolve ATRA). a) RA-sensitive cell lines demonstrated decreased proliferation (decline ≥50% compared to the control) after incubation with 0.1-10 µM ATRA. b) RA-resistant cell lines demonstrated decreased proliferation after incubation with ≥50 µM ATRA. Diagrams were plotted by using mean values from three independent repeats.
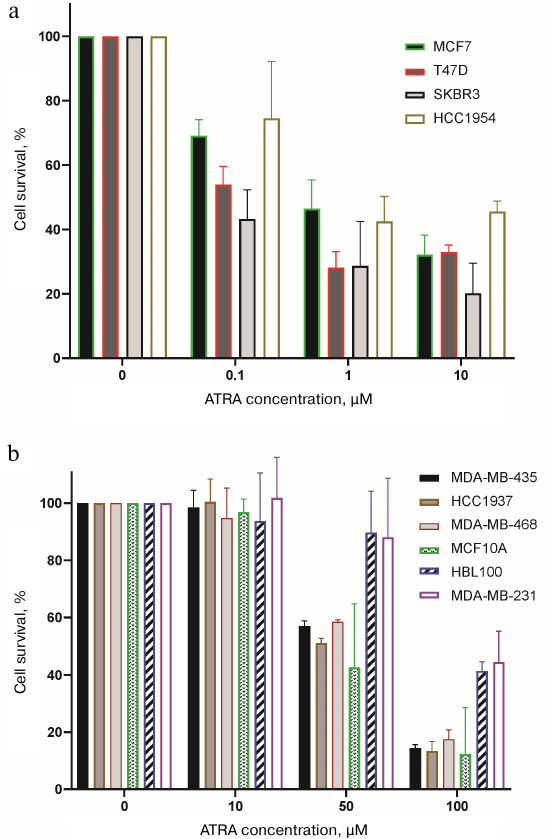
Hence, it can be concluded that RA sensitivity varies in a broad range, and the threshold level of 1-10 µM noticeably reduces cell proliferation of the RA-sensitive cells by at least 2-fold compared to the control that does not change after that, whereas proliferation of the RA-resistant cells either remained unchanged or start to decline only at 50 µM ATRA, i.e., at RA concentration markedly exceeding physiological level.
Inter-connected CRABP1 and CRABP2 production in BC cells with CRABP2 serving as a CRABP1 regulator. Next, we analyzed production of the RA-binding proteins CRABP1 and CRABP2 in the RA-sensitive and RA-resistant BC cell lines. The data demonstrated no association between the CRABP expression and RA-sensitivity. In particular, the RA-sensitive cell line SKBR3 was shown to lack CRABP1, whereas the RA-resistant lines MDA-MB-453, MDA-MB-468, and HCC1937 showed high level of this protein expression (Fig. 3a).
Fig. 3. Production of CRABP1 and CRABP2 proteins in the BC cell lines with varying RA-sensitivity. a) Percentage of live cells in each line were compared with the relevant control (the number of survived cells used as 100%) after incubation with 10 µM ATRA and related immunoblotting data (below). b) Specificity of anti-CRABP1 and anti-CRABP2 protein antibodies was assessed in the MDA-MB-231 subline derivative overexpressing CRABP1 (MDA-MB-231 CRABP1) or expressing an empty vector (MDA-MB-231 pLXSN).
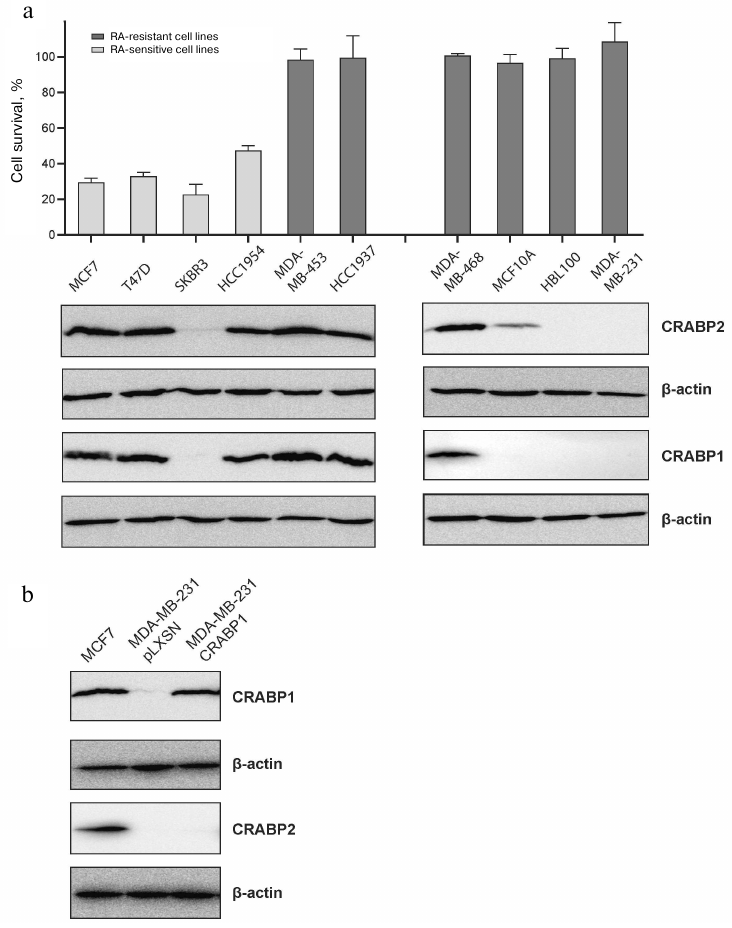
Nonetheless, it is worth noting that the most resistant lines MDA-MB-231 and HBL100, proliferation of which was not much affected even after exposure to 50 µM ATRA, did not produce CRABP1. Hence, RA-sensitivity of the BC cell lines was either unrelated to CRABP1 expression or determined by combination of factors that could involve the state of receptors ER, PR, HER2/neu and other characteristics. At the same time, the data obtained with the BC cell lines confirmed our hypothesis about correlation between the CRABP1 and CRABP2 protein levels. In particular, all cell lines exhibiting pronounced CRABP2 expression also demonstrated high level of CRABP1 production, while those displaying significantly lower CRABP2 production had almost undetectable CRABP1 levels, and the cell lines that lacked CRABP2 production also did not have CRABP1 expression. We additionally analyzed specificity of the antibodies by using the MDA-MB-231 cell line without endogenous expression of CRABP1 that was transduced with the CRABP1 coding sequence. As can be seen in Fig. 3b the overexpressed CRABP1 in the derivative subline MDA-MB-231-CRABP1 (Fig. 3b) is clearly detected with the antibodies against this protein, but this is not accompanied by upregulation of the CRABP2 expression indicating, in particular, selectivity of the antibodies to these proteins.
Taking into consideration the literature data on CRABP protein expression in the cells originating from other lineage than BC, the data obtained in our study strongly suggest existence of general correlation between CRABP1 and CRABP2 proteins that implies mutual regulation between them. It is important to note that the absence of CRABP1 was detected together with the low CRABP2 expression, but not vice versa – no cases of low CRABP1 expression and lack of CRABP2 production were observed. It is also important that the overexpressed CRABP1 in both cases (cell lines H460 and MDA-MB-231) did not change the level of CRABP2 expression. Therefore, we suggested that CRABP2 might act as a CRABP1 regulator.
To test this hypothesis, endogenous CRABP2 expression was knocked down in the four BC cell lines with varying RA-sensitivity: RA-sensitive lines MCF7 and T47D as well as RA-resistant cells MDA-MB-453 and MDA-MB-468. For this, we applied the RNA interference approach by using short hairpin RNA (shRNA) precursors against CRABP2 and cloning two shRNA (sh1CRABP2 and sh2CRABP2) sequences into the vector pLKO.puro to be further transduced into the cells via lentivirus infection followed by puromycin-based selection. Efficacy of both shRNA constructs turned out to be high allowing downregulation of the CRABP2 production compared to the control lines expressing shRNA against green fluorescence protein (shGFP), that according to densitometry analysis of immunoblotting data varied in the different cell lines from 1.6 up to 10-fold for sh1CRABP2 and from 2.5 up to 10-fold for sh2CRABP2 (Fig. 4). Analysis of the CRABP1 production in the derivative sublines demonstrated markedly lower production in all cells with silenced CRABP2 gene (Fig. 4) compared to the relevant control. It suggests that the level of CRABP1 production is regulated by CRABP2 protein.
Fig. 4. CRABP2 knockdown affects CRABP1 production in the BC cell lines. RA-sensitive (MCF7, T47D) and RA-resistant (MDA-MB-453, MDA-MB-468) BC cell line derivatives expressing short hairpin RNAs against CRABP2 mRNA (sh1CRABP2 and sh2CRABP2). Cell lines expressing short hairpin RNA against green fluorescence protein mRNA (shGFP) were used in control. Densitometry data from three independent experiments are presented.
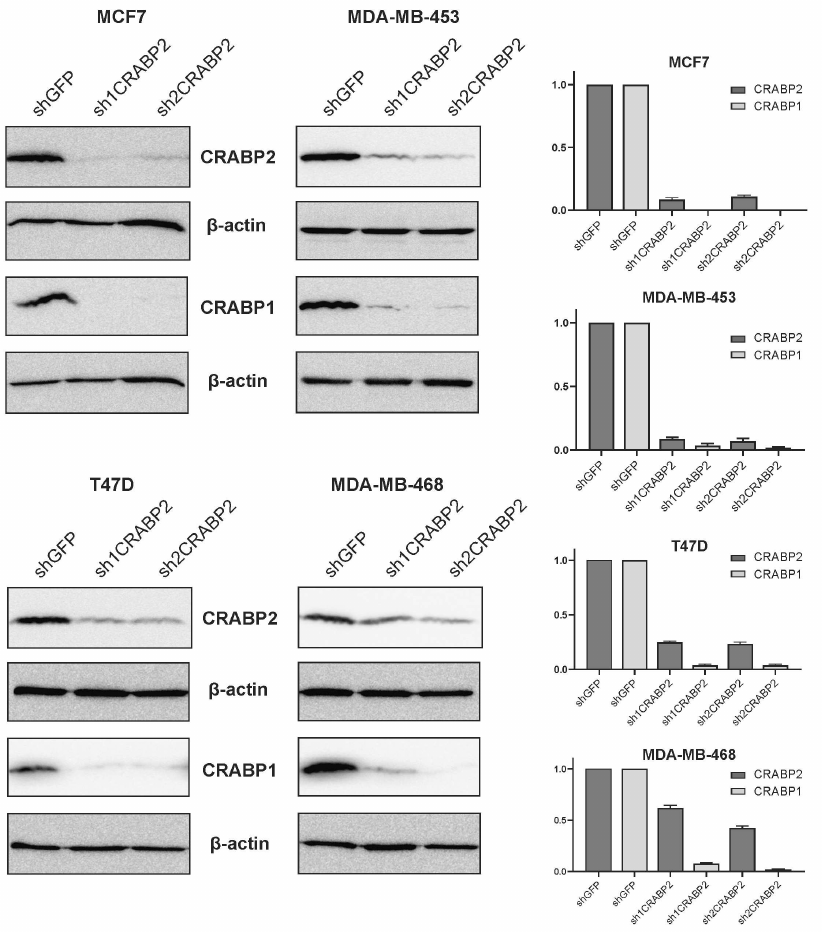
Thus, for the first time we demonstrated strong correlation between the expression of proteins CRABP1 and CRABP2 and elucidated mechanism for such interplay, wherein CRABP2 protein acts as an upstream regulator for CRABP1 protein expression. Versatility of this phenomenon demonstrates its biological significance. CRABP2-driven regulation of CRABP1 production serves as an additional mechanism for realization of the retinoic acid intracellular activity.
DISCUSSION
Formation of RA-resistance is one of the major limitations for using RA in clinical practice despite that development of the RA-based anti-tumor therapeutics both natural and synthetic has been conducted for a long time [28]. So far, the mechanisms of emerging RA-resistance remain poorly understood. CRABP1 protein may be considered as one of the possible participants in the development of RA-resistance that was proposed based upon both its potential participation in restricting RA bioavailability due to its cytoplasmic sequestration and stimulation of its catabolism by cytochrome Cyp26A1 [18], as well as due to the CRABP1 gene methylation detected in the tumors [29-33] considered as predominantly RA-resistant. At the same time, our data show that this protein in neuroblastoma cells (a classic example of the RA-sensitive tumors) is expressed at high level, so that its production correlates with the neuroblastoma differentiation level [34]. Presumably, CRABP1 may safeguard such RA-sensitive cells from the excessive amounts of RA. At the same time, a tumor-promoting role of CRABP1 in some tumors was demonstrated in our work [22] and others [11, 12, 35, 36], which was potentially associated with the competitive transport of RA to CRABP2 protein, a major mediator of the tumor-suppressor RA activity realized through RA delivery to the nuclear receptors RAR/RXR followed by activated transcription of the pro-apoptotic, pro-differentiation, and anti-proliferative genes (Fig. 5a). In other words, the tumor-promoting and tumor-suppressing (in the case of neuroblastoma) roles of CRABP1 could be related to its function of limiting RA activity. Perhaps, this fact (varying RA sensitivity of certain cancer cells) could provide explanation to the crucial contradictions in the published data regarding functional importance of CRABP1 (tumor-promoting or tumor-suppressor role) in carcinogenesis. The data showing a link between CRABP1 expression and receptor-negative BC state [15] that in turn is often related to RA-resistance in this cancer type [37-39] may indirectly indicate the CRABP1 involvement in the development of RA-resistance. We suggest that CRABP1 may facilitate acquiring of the RA-resistance by the initially RA-sensitive cells through limiting RA activity controlling transcription of the RA-sensitive genes. These limitations become unnecessary in the RA-resistant cells that may cause methylation of the CRABP1 gene. In this study we compared expression of CRABP1 protein in the cancer cell lines of various origin for the first time and found that the tumor cell lines considered as RA-resistant (three NSCLC, two glioblastoma, and three ovarian cancer cell lines) lacked CRABP1 production, whereas in all three neuroblastoma cell lines (RA-sensitive tumors) CRABP1 was detected at quite high levels. BC is an extremely heterogeneous group of tumors both in terms of morphology and molecular traits, including RA-sensitivity. To understand whether direct correlation between the CRABP1 expression and RA-sensitivity does exist, we assessed effect of RA on the proliferative activity in 9 BC cell lines and found a broad range of reactions to ATRA exposure, so that the cells were well separated into two group of cell lines with one exhibiting markedly (at least 2-fold) lower proliferation following exposure to as low as 1-10 µM ATRA (RA-sensitive) and cell lines with proliferation unaffected by exposure to 10-20 µM ATRA (RA-resistant). Comparing CRABP1 production in these cells revealed no correlation with RA-sensitivity. Although it should be mentioned that the cell lines showing maximum RA-resistance, which had unaltered proliferation after incubation with ATRA even at concentration of 50 µM, lacked CRABP1 production. The BC RA-sensitive cells were CRABP1-positive except SKBR3 cells. Overall, these ambiguous data do not allow concluding on whether CRABP1 is involved or not in the developing of RA-resistance primarily due to the fact that multiple potential mechanisms of RA-resistance could exist as has been suggested in the available publications [40]. In addition, it should be taken into consideration the aforementioned breast cancer heterogeneity that may account for the RA-resistance developing via diverse molecular scenarios [41].
Discovery of the 100% correlation between CRABP1 and CRABP2 expression is probably the most prominent result of this study. It should be emphasized that such phenomenon was confirmed in all cancer cell types regardless of their origin and RA-sensitivity thereby indicating its biological significance. Our data allowed suggesting that production of CRABP proteins is functionally inter-connected so that CRABP2 seems to act as an upstream regulator. Previously, this issue has not been investigated. The data presented in a single report examining both CRABP proteins simultaneously with assessing their functional role demonstrated a rather competitive interaction between CRABP1 and CRABP2 proteins. It was assumed that CRABP1 limits CRABP2 activity with regard to RA nuclear activity by sequestering RA in the cytoplasm [15]. However, it implies functional competition rather than regulating protein quantity. In this context, it is worth noting our previous data on the correlated mRNA and protein expression of both CRABP1 and CRABP2 in the lung adenocarcinoma samples. Moreover, the Spearman correlation coefficient peaked in the high- or middle-grade NSCLCs, but correlation was absent in the low-grade cancer cells [21]. Indirectly, it suggests that interplay between the CRABP proteins exists implying some regulatory mechanism that seems to be lost during tumor progression and cell dedifferentiation.
The current study confirmed existence of such regulatory axis as well as demonstrated that CRABP2 acts as CRABP1 regulator. So, how such CRABP2-dependent regulation of CRABP1 activity might occur? Several scenarios may be proposed (Fig. 5b).
Fig. 5. Potential mechanisms underlying effects of CRABP2 on intracellular CRABP1 protein level. a) Schematic representation of RA functional activity. CRABP2 protein ensures RA nuclear trafficking and its transfer to cognate nuclear RAR receptors. FABP5 protein delivers RA to the nuclear receptor PPARβ/δ. Formation of the complex between RA and RXR-containing heterodimer receptors stimulates interaction with retinoic acid response elements (RARE) within the gene promoters as well as recruitment of co-activators and transcription activation. Similar activity is presumably exerted by CRAPB1 protein, but compared to CRAPB2, it forms no complex with RA receptors – it is believed that in this case the RA-CRABP1 complex dissociation precedes RA transfer to the nuclear receptors. b) Hypothetic scheme describing CRABP2-dependent regulation of CRABP1 production. Similar regulation may be realized via the CRABP2-dependent activation of CRABP1 transcription, CRABP2-dependent stabilization of CRABP1 mRNA involving members from the Hu-protein group (e.g., HuR), as well as potentially via the negatively regulated proteolytic CRABP1 degradation.
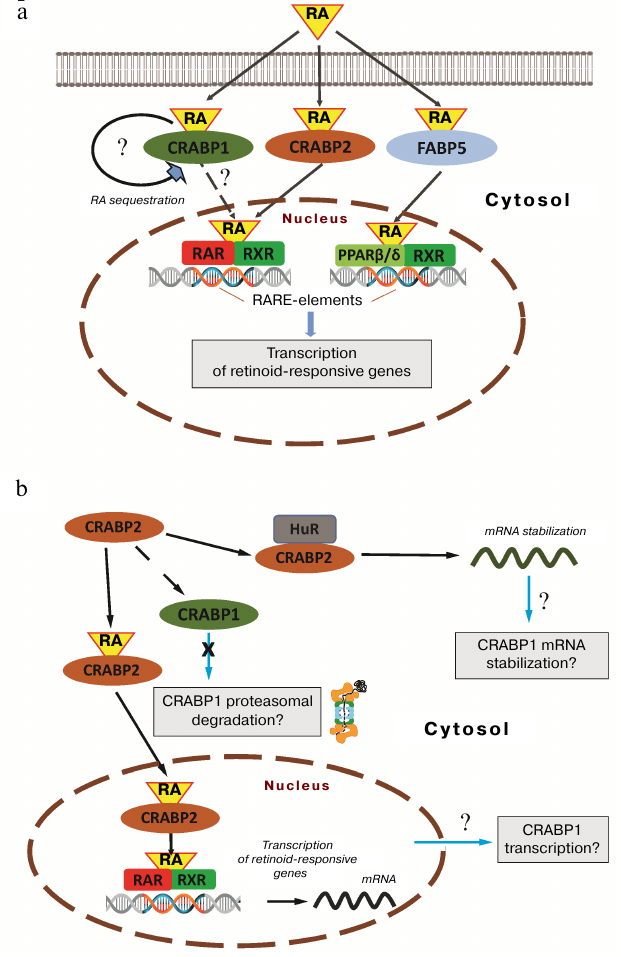
The first scenario implies directly regulated expression, when the CRABP2-dependent RA delivery to nuclear receptors results in upregulated CRABP1 gene transcription, which promoter carries RA-responsive element. Apart from the transcriptional transactivation, a mechanism of CRABP2-dependent CRABP1 transcript stabilization may be also suggested, which is based upon the CRABP2 activity unrelated to RA shown in several studies and executed via interaction with the members of the Hu-protein group belonging to the ELAV (embryonic lethal abnormal vision) RNA-binding proteins. Such proteins are mainly involved in the post-transcriptionally regulated gene expression as well as in stabilizing mRNA and some other RNA types (e.g., microRNA) [42]. CRABP2 apparently targets several proteins from that group including HuD and HuB [9, 10, 43]. Interaction of CRABP2 with Hu-proteins results in their subsequent activation followed by increased production of a whole set of the regulatory and signaling proteins. Interestingly, HuR, in turn, facilitates CRABP2 nuclear trafficking, thereby stimulating its activity to deliver RA to the nuclear receptors. In addition, it may also be assumed that the CRABP2-dependent regulation occurs not at the transcriptional level or post-transcriptionally regulated mRNA, but rather due to proteolytical degradation of CRABP1 activated in the absence of CRABP2. The regulatory mechanisms employed in this scenario have been shown for the functionally associated or related proteins. It could be illustrated by the example of flotillin-1 and flotillin-2, for which it was observed many times that suppression of either of isoforms resulted in the reduced level of its counterpart [44-46]. All the aforementioned potential mechanisms will be further investigated and could provide deeper insights into understanding of the fundamental processes underlying retinoic acid intracellular activity.
Funding. This study was financially supported by the Russian Foundation for Basic Research (project no. 19-015-00027A).
Ethics declarations. The authors declare no conflict of interest. This article does not contain any studies with human participants or animals performed by any of the authors.
REFERENCES
1.Connolly, R. M., Nguyen, N. K., and Sukumar, S.
(2013) Molecular pathways: current role and future directions of the
retinoic acid pathway in cancer prevention and treatment, Clin.
Cancer Res., 19, 1651-1959, doi:
10.1158/1078-0432.CCR-12-3175.
2.Schenk, T., Stengel, S., and Zelent, A. (2014)
Unlocking the potential of retinoic acid in anticancer therapy, Br.
J. Cancer, 111, 2039-2045, doi: 10.1038/bjc.2014.412.
3.Chevkina, E. M., and Favorskaya, I. A. (2015) CRABP
proteins – relatives or namesakers? [in Russian], Uspekhi Mol.
Onkol., 2, 6-16, doi: 10.17650/2313-805X.2015.2.2.6-16.
4.Tchevkina, E. M. (2017) Retinoic acid binding
proteins and cancer: similarity or polarity? Cancer Ther. Oncol.
Int. J., 8, 555733, doi: 10.19080/ctoij.2017.08.555733.
5.Sussman, F., and De Lera, A. R. (2005) Ligand
recognition by RAR and RXR receptors: binding and selectivity, J.
Med. Chem., 48, 6212-6219, doi: 10.1021/jm050285w.
6.Schug, T. T., Berry, D. C., Shaw, N. S., Travis, S.
N., and Noy, N. (2007) Opposing effects of retinoic acid on cell growth
result from alternate activation of two different nuclear receptors,
Cell, 129, 723-733, doi: 10.1016/j.cell.2007.02.050.
7.Schug, T. T., Berry, D. C., Toshkov, I. A., Cheng,
L., Nikitin, A. Y., and Noy, N. (2008) Overcoming retinoic
acid-resistance of mammary carcinomas by diverting retinoic acid from
PPARβ/δ to RAR, Proc. Natl. Acad. Sci. USA,
105, 7546-7551, doi: 10.1073/pnas.0709981105.
8.Liu, R. Z., Graham, K., Glubrecht, D. D., Germain,
D. R., Mackey, J. R., and Godbout, R. (2011) Association of FABP5
expression with poor survival in triple-negative breast cancer:
implication for retinoic acid therapy, Am. J. Pathol.,
178, 997-1008, doi: 10.1016/j.ajpath.2010.11.075.
9.Vreeland, A. C., Levi, L., Zhang, W., Berry, D. C.,
and Noy, N. (2014) Cellular retinoic acid-binding protein 2 inhibits
tumor growth by two distinct mechanisms, J. Biol. Chem.,
289, 34065-34073, doi: 10.1074/jbc.M114.604041.
10.Vreeland, A. C., Yu, S., Levi, L., de Barros
Rossetto, D., and Noy, N. (2014) Transcript stabilization by the
RNA-binding protein HuR is regulated by cellular retinoic acid-binding
protein 2, Mol. Cell. Biol., 34, 2135-2146, doi:
10.1128/mcb.00281-14.
11.Mallikarjuna, K., Sundaram, C. S., Sharma, Y.,
Deepa, P. R., Khetan, V., et al. (2010) Comparative proteomic analysis
of differentially expressed proteins in primary retinoblastoma tumors,
Proteom. Clin. Appl., 4, 449-463, doi:
10.1002/prca.200900069.
12.Liu, R. Z., Li, S., Garcia, E., Glubrecht, D. D.,
Yin Poon, H., et al. (2016) Association between cytoplasmic CRABP2,
altered retinoic acid signaling, and poor prognosis in glioblastoma,
Glia, 64, 963-976, doi: 10.1002/glia.22976.
13.Dong, D., Ruuska, S. E., Levinthal, D. J., and
Noy, N. (1999) Distinct roles for cellular retinoic acid-binding
proteins I and II in regulating signaling by retinoic acid, J. Biol.
Chem., 274, 23695-23698, doi: 10.1074/jbc.274.34.23695.
14.Blaese, M. A., Santo-Hoeltje, L., and Rodemann,
H. P. (2003) CRABP I expression and the mediation of the sensitivity of
human tumour cells to retinoic acid and irradiation, Int. J. Radiat.
Biol., 79, 981-991, doi: 10.1080/09553000310001632949.
15.Liu, R. Z., Garcia, E., Glubrecht, D. D., Poon,
H. Y., Mackey, J. R., and Godbout, R. (2015) CRABP1 is associated with
a poor prognosis in breast cancer: adding to the complexity of breast
cancer cell response to retinoic acid, Mol. Cancer, 14,
129, doi: 10.1186/s12943-015-0380-7.
16.Fiorella, P. D., and Napoli, J. L. (1991)
Expression of cellular retinoic acid binding protein (CRABP) in
Escherichia coli. Characterization and evidence that
holo-CRABP is a substrate in retinoic acid metabolism, J. Biol.
Chem., 266, 16572-16579.
17.Boylan, J. F., and Gudas, L. J. (1992) The level
of CRABP-I expression influences the amounts and types of all-
trans-retinoic acid metabolites in F9 teratocarcinoma stem cells, J.
Biol. Chem., 267, 21486-21491.
18.Won, J. Y., Nam, E. C., Yoo, S. J., Kwon, H. J.,
Um, S. J., et al. (2004) The effect of cellular retinoic acid binding
protein-I expression on the CYP26-mediated catabolism of all-trans
retinoic acid and cell proliferation in head and neck squamous cell
carcinoma, Metab. Clin. Exp., 53, 1007-1012, doi:
10.1016/j.metabol.2003.12.015.
19.Delektorskaya, V. V., Komel’kov, A. V.,
Zborovskaya, I. B., Enikeev, A. D., Safronova, V. M., and Chevkina, E.
M. (2017) Nuclear localization of cellular retinoic acid-binding
protein 1 (Crabp1) is associated with malignancy level in lung
neuroendocrine tumors [in Russian], Voprosy Onkologii,
63, 886-893.
20.Gaub, M. P., Lutz, Y., Ghyselinck, N. B.,
Scheuer, I., Pfister, V., et al. (1998) Nuclear detection of cellular
retinoic acid binding proteins I and II with new antibodies, J.
Histochem. Cytochem., 46, 1103-1111, doi:
10.1177/002215549804601002.
21.Favorskaya, I., Kainov, Y., Chemeris, G.,
Komelkov, A., Zborovskaya, I., and Tchevkina, E. (2014) Expression and
clinical significance of CRABP1 and CRABP2 in non-small cell lung
cancer, Tumor Biol., 35, 10295-10300, doi:
10.1007/s13277-014-2348-4.
22.Kainov, Y., Favorskaya, I., Delektorskaya, V.,
Chemeris, G., Komelkov, A., et al. (2014) CRABP1 provides high
malignancy of transformed mesenchymal cells and contributes to the
pathogenesis of mesenchymal and neuroendocrine tumors, Cell
Cycle, 13, 1530-1539, doi: 10.4161/cc.28475.
23.Rossetti, S., and Sacchi, N. (2019) 3D mammary
epithelial cell models: a goldmine of dcis biomarkers and morphogenetic
mechanisms, Cancers, 11, 130, doi:
10.3390/cancers11020130.
24.Garattini, E., Bolis, M., Garattini, S. K.,
Fratelli, M., Centritto, F., et al. (2014) Retinoids and breast cancer:
from basic studies to the clinic and back again, Cancer Treat.
Rev., 40, 739-749, doi: 10.1016/j.ctrv.2014.01.001.
25.Coyle, K. M., Dean, C. A., Thomas, M. L.,
Vidovic, D., Giacomantonio, C. A., et al. (2018) DNA methylation
predicts the response of triple-negative breast cancers to all-trans
retinoic acid, Cancers, 10, 397, doi:
10.3390/cancers10110397.
26.Centritto, F., Paroni, G., Bolis, M., Garattini,
S. K., Kurosaki, M., et al. (2015) Cellular and molecular determinants
of all‐trans retinoic acid sensitivity in breast cancer: luminal
phenotype and RARα expression, EMBO Mol. Med., 7,
950-972, doi: 10.15252/emmm.201404670.
27.Bolis, M., Garattini, E., Paroni, G., Zanetti,
A., Kurosaki, M., et al. (2017) Network-guided modeling allows
tumor-type independent prediction of sensitivity to all-trans-retinoic
acid, Ann. Oncology, 28, 611-621, doi:
10.1093/annonc/mdw660.
28.Coyle, K. M., Sultan, M., Thomas, M. L.,
Vaghar-Kashani, A., and Marcato, P. (2013) Retinoid signaling in cancer
and its promise for therapy, J. Carcinogen. Mutagen., doi:
10.4172/2157-2518.s7-006.
29.Miyake, T., Ueda, Y., Matsuzaki, S., Miyatake,
T., Yoshino, K., et al. (2011) CRABP1-reduced expression is associated
with poorer prognosis in serous and clear cell ovarian adenocarcinoma,
J. Cancer Res. Clin. Oncol., 137, 715-722, doi:
10.1007/s00432-010-0930-8.
30.Tanaka, K., Imoto, I., Inoue, J., Kozaki, K.,
Tsuda, H., et al. (2007) Frequent methylation-associated silencing of a
candidate tumor-suppressor, CRABP1, in esophageal squamous-cell
carcinoma, Oncogene, 26, 6456-6468, doi:
10.1038/sj.onc.1210459.
31.Lind, G. E., Kleivi, K., Meling, G. I., Teixeira,
M. R., Thiis-Evensen, E., et al. (2006) ADAMTS1, CRABP1, and NR3C1
identified as epigenetically deregulated genes in colorectal
tumorigenesis, Cell. Oncol., 28, 259-272, doi:
10.1155/2006/949506.
32.Wu, Q., Lothe, R. A., Ahlquist, T., Silins, I.,
Tropé, C. G., et al. (2007) DNA methylation profiling of ovarian
carcinomas and their in vitro models identifies HOXA9, HOXB5,
SCGB3A1, and CRABP1 as novel targets, Mol. Cancer, 6, 42,
doi: 10.1186/1476-4598-6-45.
33.Wang, F., Yang, Y., Fu, Z., Xu, N., Chen, F., et
al. (2014) Differential DNA methylation status between breast
carcinomatous and normal tissues, Biomed. Pharmacother.,
68, 699-707, doi: 10.1016/j.biopha.2014.07.014.
34.Stroganova, A. M., Chemeris, G. Yu., Chevkina, E.
M., Senderovich, A., Karseladze, A. I. (2016) CRABP protein 1 and its
role in the process of differentiation neuroblastoma, Vestnik RONTs
im. N. N. Blokhina, 27, 157-163.
35.Bertucci, F., Houlgatte, R., Benziane, A.,
Granjeaud, S., Adélaïde, J., et al. (2000) Gene expression
profiling of primary breast carcinomas using arrays of candidate genes,
Hum. Mol. Genet., 9, 2981-2991, doi:
10.1093/hmg/9.20.2981.
36.Tsibris, J. C. M., Segars, J., Coppola, D., Mane,
S., Wilbanks, G. D., et al. (2002) Insights from gene arrays on the
development and growth regulation of uterine leiomyomata, Fertil.
Steril., 78, 114-121, doi:
10.1016/S0015-0282(02)03191-6.
37.Fontana, J. A. (1992) Responses to retinoic acid
of tamoxifen-sensitive and -resistant sublines of human breast cancer
cell line MCF-7, Cancer Res., 52, 6164-6167.
38.Fontana, J. A. (1987) Interaction of retinoids
and tamoxifen on the inhibition of human mammary carcinoma cell
proliferation, Pathobiology, 55, 136-144, doi:
10.1159/000163409.
39.Van der Leede, B. J. M., Folkers, G. E., van den
Brink, C. E., van der Saag, P. T., and van der Burg, B. (1995) Retinoic
acid receptor α1 isoform is induced by estradiol and confers
retinoic acid sensitivity in human breast cancer cells, Mol. Cell.
Endocrinol., 109, 77-86, doi:
10.1016/0303-7207(95)03487-R.
40.Chlapek, P., Slavikova, V., Mazanek, P., Sterba,
J., and Veselska, R. (2018) Why differentiation therapy sometimes
fails: molecular mechanisms of resistance to retinoids, Int. J. Mol.
Sci., 19, 132, doi: 10.3390/ijms19010132.
41.Tari, A. M., Lim, S. J., Hung, M. C., Esteva, F.
J., and Lopez-Berestein, G. (2002) Her2/neu induces all-trans retinoic
acid (ATRA) resistance in breast cancer cells, Oncogene,
21, 5224-5232, doi: 10.1038/sj.onc.1205660.
42.Wang, J., Guo, Y., Chu, H., Guan, Y., Bi, J., and
Wang, B. (2013) Multiple functions of the RNA-binding protein HuR in
cancer progression, treatment responses and prognosis, Int. J. Mol.
Sci., 14, 10015-10041, doi: 10.3390/ijms140510015.
43.Gupta, A., Williams, B. R. G., Hanash, S. M., and
Rawwas, J. (2006) Cellular retinoic acid-binding protein II is a direct
transcriptional target of MycN in neuroblastoma, Cancer Res.,
66, 8100-8108, doi: 10.1158/0008-5472.CAN-05-4519.
44.Babuke, T., Ruonala, M., Meister, M., Amaddii,
M., Genzler, C., et al. (2009) Hetero-oligomerization of
reggie-1/flotillin-2 and reggie-2/flotillin-1 is required for their
endocytosis, Cell. Signal., 21, 1287-1297, doi:
10.1016/j.cellsig.2009.03.012.
45.Frick, M., Bright, N. A., Riento, K., Bray, A.,
Merrified, C., and Nichols, B. J. (2007) Coassembly of flotillins
induces formation of membrane microdomains, membrane curvature, and
vesicle budding, Curr. Biol., 17, 1151-1156, doi:
10.1016/j.cub.2007.05.078.
46.Solis, G. P., Hoegg, M., Munderloh, C., Schrock,
Y., Malaga-Trillo, E., et al. (2007) Reggie/flotillin proteins are
organized into stable tetramers in membrane microdomains, Biochem.
J., 403, 313-322, doi: 10.1042/BJ20061686.