The Structure of the Potato Virus A Particles Elucidated by Small Angle X-Ray Scattering and Complementary Techniques
Eleonora V. Shtykova1,a, Maxim V. Petoukhov1, Natalia V. Fedorova2, Alexander M. Arutyunyan2, Eugene V. Skurat3, Larisa V. Kordyukova2 , Andrey V. Moiseenko3, and Alexander L. Ksenofontov1,2,b*
1Shubnikov Institute of Crystallography of Federal Scientific Research Centre “Crystallography and Photonics” of the Russian Academy of Sciences, 119333 Moscow, Russia2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119234 Moscow, Russia
3Faculty of Biology, Lomonosov Moscow State University, 119234 Moscow, Russia
* To whom correspondence should be addressed.
Received August 26, 2020; Revised September 27, 2020; Accepted September 27, 2020
Potato virus A (PVA) protein coat contains on its surface partially unstructured N-terminal domain of the viral coat protein (CP), whose structural and functional characteristics are important for understanding the mechanism of plant infection with this virus. In this work, we investigated the properties and the structure of intact PVA and partially trypsinized PVAΔ32 virions using small-angle X-ray scattering (SAXS) and complimentary methods. It was shown that after the removal of 32 N-terminal amino acids of the CP, the virion did not disintegrate and remained compact, but the helical pitch of the CP packing changed. To determine the nature of these changes, we performed ab initio modeling, including the multiphase procedure, with the geometric bodies (helices) and restoration of the PVA structure in solution using available high-resolution structures of the homologous CP from the PVY potyvirus, based on the SAXS data. As a result, for the first time, a low-resolution structure of the filamentous PVA virus, both intact and partially degraded, was elucidated under conditions close to natural. The far-UV circular dichroism spectra of the PVA and PVAΔ32 samples differed significantly in the amplitude and position of the main negative maximum. The extent of thermal denaturation of these samples in the temperature range of 20-55°C was also different. The data of transmission electron microscopy showed that the PVAΔ32 virions were mostly rod-shaped, in contrast to the flexible filamentous particles typical of the intact virus, which correlated well with the SAXS results. In general, structural analysis indicates an importance of the CP N-terminal domain for the vital functions of PVA, which can be used to develop a strategy for combating this plant pathogen.
KEY WORDS: potyviruses, coat protein, potato virus A, N-terminal disordered domains, small-angle X-ray scattering, structural modelingDOI: 10.1134/S0006297921020115
Abbreviations: CP, coat protein; PVA, potato virus A; SAXS, small angle X-ray scattering; TEM, transmission electron microscopy.
INTRODUCTION
Potyviruses represent an important group of plant viruses that cause significant economic losses in agriculture. Flexible filamentous virions of the potyviruses display helical symmetry with an average particle length of 700 nm and diameter of 11-14 nm. It was shown with the help of various predictive and experimental approaches that the coat protein (CP) of the potato virus A (PVA) – typical representative of potyviruses – has a significant portion of unstructured regions [1]. Later, this fact was directly conformed by cryoelectron microscopy for other representatives of potyviruses, such as watermelon mosaic virus (WMV) [2], potato virus Y (PVY) [3], and turnip mosaic virus (TuMV) [4]. It was demonstrated that the unstructured fragments of the N-terminal domains in the potyviral CPs are located on the surface of virions and interact with the neighboring subunits. It is commonly accepted now that the protein unstructured regions play a key role in various biochemical processes [5]. We have observed previously that the N-terminal domains of the PVA CP participate in the assembly of the virus-like particles (VLPs); it was shown that the subunits of this protein did not exist as individual molecules in solution, and part of their unstructured segments acquired the β-structure [6, 7]. Mild trypsinization of intact virions of potyviruses results in the formation of stable degraded particles lacking the N-terminal domain [8]. In order to clarify the role of the N-terminal domains of potyviral CPs, a number of studies investigated the process of VLP assembly from the truncated mutant CPs. It was found that for some representatives of potyviruses, deletion of 20-50 N-terminal residues prevented the assembly of the VLPs [9, 10], while this was not the case for the PVY [3]. At the same time, in the case of CP from tobacco etch virus (TEV), the assembly was disrupted only upon deletion of 120 amino acid residues [11]. The role of the CP N-domains in the cell-to-cell transmission of potyvirus virions and in their transmission by mites was also demonstrated [10].
The high-resolution structure of the PVA has not been obtained yet. In this study, we for the first time characterized in detail the structure of the PVA particles by modeling intact and trypsinized virions based on the data of small angle X-ray scattering (SAXS). This low-resolution structural technique provides resolution of ~1-2 nm and can be used for investigating the structure of biological molecules in solution under conditions close to natural [12]. The SAXS technique has been developed actively due to the emergence of novel methods of analysis and interpretation of the SAXS data and computer modeling [13]. Previously, we characterized the PVA VLPs assembled from the isolated free CP, as well as their structure and dissociation in different aqueous saline solutions [7, 8]. Recently, we applied this technique for solving the low-resolution structure of the helical rod-shaped tobacco mosaic virus (TMV) and of the stable repolymerized TMV CP aggregates, the so-called stacked discs [14]. Here, we found that the observed differences in the structural characteristics of the intact PVA and partially degraded PVAΔ32 virions correlated with the observed differences of their optical properties. The obtained results not only indicate the significance of the CP N-terminal domains for the vital functions of PVA, but also could facilitate the development of strategies for fighting viral infections in plants, as the unstructured CP N-terminal region could be a target for the virus deactivation.
MATERIALS AND METHODS
Preparation of intact and degraded PVA virions. An isolate of PVA virions was purified as described previously [8]. Briefly, young Nicotiana benthamiana plants were infected with a PVA preparation. After 18-21 days, leaves with the symptoms of viral infection were collected and homogenized in a Waring blender with three volumes of 50 mM phosphate buffer, pH 7.7, supplemented with 0.01% β-mercaptoethanol. Following low-speed centrifugation (LSC) at 8,000g for 20 min at 8°C, the supernatant was stirred for 1 h at 4°C with Triton X-100 (1% vol/vol). Next, 1/5 volume of chloroform was added to the LSC supernatant. Polyethylene glycol 6000 and NaCl were then added to the mixture to the concentration of 5 and 1.2%, respectively, and the mixture was stirred at 4°C for 1.5 h. The formed precipitate containing the virus was separated by LSC at 12,000g for 20 min at 8°C and dissolved in 50 mM phosphate buffer (pH 7.8) supplemented with 0.1 mM phenylmethylsulfonyl fluoride. Next, the preparation was subjected to high-speed centrifugation at 100,000g for 1.5 h at 5°C, and the precipitate was resuspended in 50 mM phosphate buffer (pH 7.8).
Degraded virions (PVAΔ32) were prepared by treating the intact virus with trypsin at an enzyme/substrate ratio of 1 : 500 for 15 min at room temperature. The reaction was stopped by addition of phenylmethylsulfonyl fluoride to the concentration of 1 mM. The purity of the sample was tested by sodium dodecyl sulfate (SDS) electrophoresis in 15% polyacrylamide gel (PAAG) according to the Laemmli procedure [15] in a Mini-PROTEAN 3 Cell (Bio-Rad, USA). The gels were stained with 0.22% Coomassie G-250 (Serva, Germany).
Determination of the virus concentration. The concentration of the viral samples was determined with a UV-2600 spectrophotometer (Hitachi, Japan) from the absorption spectra in the UV range (240-340 nm). Because of a large contribution of scattering to the absorption of virions (30-40%), true absorption spectra (E) of the particle suspension were calculated in the range of 320-338 nm using the extrapolation technique [16]. The absorption coefficient of the virus preparation was taken as E0.1%260 = 2.4.
Circular dichroism (CD) spectra. CD spectra were recorded in 5 mM phosphate buffer (pH 7.8) at 20°C in 1-2-mm cuvettes using a Chirascan circular dichrograph (Applied Photophysics, United Kingdom). The sample concentration was 50-100 µg/ml. The CD spectra were recorded at a scanning speed of 0.5-1.0 nm/s at 185-250 nm with background subtraction and processed using a standard software supplied with the instrument. The CD values were expressed in the molar ellipticity units [Θ] ([Θ] = 3300×Δε), where Δε – molar coefficient of dichroic absorption for the preparations calculated per mole of amino acid residues. Calculations were performed using the equation Δε = ΔD/(c×l), where ΔD is the measured dichroism value; c is the protein concentration (in moles of amino acid residues); l is the optical path length in centimeters. The average molecular mass of an amino acid residue was taken as 110 g/mol. For thermoanalysis, each preparation was heated in a dichrograph cell from 20 to 70°C at a rate of 1°C per minute. Each new spectrum was recorded after the temperature was increased by 5°C. The recorded spectra were processed as described above.
Transmission electron microscopy. Samples were examined with a JEM-2100 electron microscope, 200 kV (JEOL, Japan) equipped with an electron source LaB6 using instrumentation of the Unique Scientific Setup 3D-EMS at the Lomonosov Moscow State University (ID no. RFMEFI61919X0014). The images were taken with a Gatan Ultrascan 1000XP 2k CCD camera (Gatan, USA) with parallel illumination and out-focusing in the range of 1 to 2 µm and processed with the Gatan Digital Micrograph software.
SAXS experiment and data processing. Examination of the protein structure using small angle X-ray scattering was carried on a Petra III synchrotron (DESY, Hamburg, Germany) at the beamline P12 equipped with an automatic sampler and a two-dimensional Pilatus 2M detector (DECTRIS, Switzerland). The intensity of scattering I(s) was measured in the range of wave vectors 0.08 < s < 7 nm–1, where s = (4πsinθ)/λ is the scattering vector; 2θ is the angle of scattering; and λ = 0.124 nm is the emission wavelength [17]. For each sample, 50 experimental scattering curves were recorded to control possible radiation damage. No radiation damage was observed.
SAXS measurements were conducted in 15 mM phosphate buffer (pH 7.8) containing 0.05 M NaCl at 10°C at several different protein concentrations (2.0 to 5.0 mg/ml) to account for possible concentration dependence. No dependence of SAXS curves on the concentration of tested samples was observed.
Initial processing of the scattering curves was done using the PRIMUS software [18]. The averaging of the experimental scattering curves from samples and respective buffer was conducted with the same program followed by subtraction of the buffer scattering from the SAXS curves from the samples. Next, the most informative part of the SAXS curves in the range of wave vectors 0.08 < s < 2.7 nm–1 was used for the structural analysis and modeling.
The ambiguity of the shape reconstruction for a given scattering profile was estimated with the AMBIMETER program [19].
Analysis of the Bragg peaks on the SAXS curves was carried out with the PEAK program [18]. The size of the crystallinity region L was determined from the width at the half-maximum intensity of the first Bragg peak at the angle 2θ1:
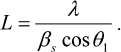 (1)
(1)The degree of the sample disordering Δ/d1 was determined as:
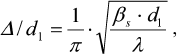 (2)
(2)where d1 = 2π/s1 is the period of the structure, interplane distance; βs is the full width at the half-maximum of scattering intensity (in radians) observed at the scattering angle 2θ1 corresponding to the wave vector s1; and Δ is the standard deviation between the closest regularly packed motifs.
The GNOM program was used to build the distance distribution functions p(r) required for the reconstruction of the virion shape in solution form the SAXS data [20]. The distance distribution functions p(r) were determined with the indirect Fourier transform of the scattering intensity in accordance with the integral equation:
where I(s) is the scattering intensity, with the maximal particle size (Dmax) determined under condition of p(r) = 0 at r > Dmax.
The low-resolution shape of the PVA virion was reconstructed ab initio with the DAMMIN program [21], which uses annealing simulation algorithm by minimizing χ2 discrepancy with the experimental data:
where N is the number of experimental points; Iexp(sj) and σ(sj) are experimental intensities and their errors; Icalc(sj) is the intensity calculated for the model; c is the scaling factor.
Using more universal ab initio approach, the model of the virus was obtained by simultaneous construction of both the general shape and of the structure of the virion individual components (protein and RNA). The two-component (two-phase) model was reconstructed by approximation of the SAXS curve for the viral particle using the MONSA program [21]. The program presents a particle as a collection of N >> 1 densely packed beads inside the search volume prescribed by the user. To describe the general and the internal structure of a complex particle, each bead can be assigned either to the solvent (index = 0), or to one of the components (in our case, index = 1 corresponded to the CP and index = 2 corresponded to the viral RNA). Hence, the complex between the protein and the RNA is presented with a low resolution as two phases, and the structure is described by a string of length N containing phase index for each bead (0, 1, or 2).
Rigid body modelling of the PNA particle was performed using the MASSHA program [22]. The PVYΔ43 fragment of the potato virus Y (PDB ID: 6HXX) was selected as a modeling unit based on the fact that the primary sequences of the PVA and PVY CPs are very similar (64% of identical and 91% similar amino acid residues), excluding the N-terminal fragments. The theoretical scattering intensity from the reconstructed models was calculated with the CRYSOL program [23].
Alignment of amino acid sequences for the representatives of the potyvirus genus was performed on the Expasy server using the LALIGN algorithm [24]. Quantitative estimation of the sequence similarity was conducted by pairwise alignment of the CP core parts lacking 30-32 N-terminal residues.
RESULTS AND DISCUSSION
Electrophoretic analysis of viral samples. PVA CP displays abnormally low electrophoretic mobility [1] corresponding to ~35 kDa (Fig. 1, lanes 2-4), while its molecular mass (M) is 30.4 kDa, which can be explained by the presence of unstructured segments in the protein molecule. Trypsinization of the intact virions under mild conditions (enzyme/substrate ratio 1 : 500, 15-min incubation) resulted in the disappearance of the band corresponding to the original protein; the major part of the particles in this case (about 95% of total mass) was represented by the band corresponding to M ~ 27 kDa (Fig. 1, lanes 5 and 6), which was consistent with the molecular mass of the truncated CP lacking the N-terminal fragment. Recently, almost complete disappearance of the peak with m/z = 30.4 kDa (original CP) and emergence of the major 27.12-kDa peak corresponding to the protein lacking 32 N-terminal residues (ABKΔ32) was demonstrated by mass spectrometry [8]. Hence, the trypsinized PVAΔ32 CP protein indeed demonstrates electrophoretic mobility corresponding to original CP protein with truncated N-terminal sequence, thus indirectly confirming the removal of the N-terminal fragment.
Fig. 1. Electrophoretic analysis of intact PVA virions (lanes 2-4) and products of their limited trypsinolysis (lanes 5 and 6): 1 µg (lanes 4 and 5), 5 µg (lane 2), and 10 µg (lanes 3 and 6) per lane. Molecular mass (kDa) markers are indicated on the left (lane 1).
Analysis of virion secondary structure based on CD spectra. Changes in the secondary structure of the PVA CP were examined using CD spectra. The CD spectrum of the intact PVA virions in the far-UV region is characterized with the negative maximum [θ]max at 203 nm (Fig. 2a, curve 2), although it is known that for the majority of plant viruses, this maximum is located at ~208 nm. It is likely that this anomaly could be explained by an unusually high content of unstructured fragments in the CP [1]. To our best knowledge, PVA is the only plant virus with such shift of the major negative maximum of the CD spectrum in the far-UV region. A moderate shift of the negative maximum [θ]max towards 205 nm was observed for the preparations of PVAΔ32 virions, and the intensity of this maximum was significantly lower than in the case of intact virions (–10600° and –25000°, respectively). Presumably, the content of unstructured segments decreased with the removal of the N-domain from the CP in PVAΔ32 (Fig. 2a, curve 1).
Fig. 2. a) CD spectra of trypsinized (1) and intact (2) PVA virions; b) thermal denaturation of trypsinized (1) and intact (2) PVA preparations based on the CD spectra. The temperature dependence of the negative maximum [θ]max intensity is presented.
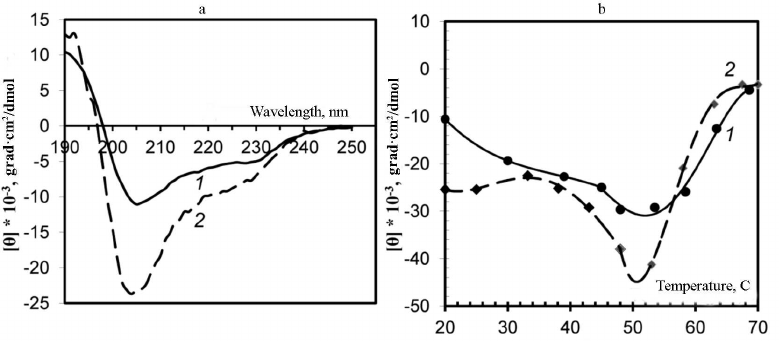
The degree of thermal denaturation also differed for the intact and trypsin-degraded PVA virions (Fig. 2b). It was shown previously that melting of intact PVA virions occurs at m.t. ≈ 55°C [1]. In this study, we monitored changes of the CD spectrum negative maxima [θ]max during heating. In the temperature range of 20-55°C, a gradual increase in the [θ]max intensity to –45,000° and –29,600° was observed for the intact PVA and PVAΔ32, respectively (Fig. 2b). It is likely that the CP in PVAΔ32 was denatured by heating to a lesser degree and the PVAΔ32 viral particles were more structured than the intact virions.
Analysis of SAXS data and structure modeling. Two main features were identified in the experimental SAXS curves of both intact PVA and trypsinized PVAΔ32 (Fig. 3, curves 1 and 2, respectively): firstly, the well-pronounced dependence of scattering on the virion shape and, secondly, the presence of the Bragg peak in the range of s from 1.5 to 2.0 nm–1 on each small-angle scattering curve.
The virion shape was determined form the initial part of the SAXS curves within the range of wave vectors s from 0.08 to 1.25 nm–1. In this interval, locations of the minimum (smin = 0.61 ± 0.01 nm–1) and the maximum (smax = 0.81 ± 0.01 nm–1) on both curves coincided, which indicates the similarity of shapes of the intact and PVAΔ32 virions. However, the SAXS curve for the intact virions is smoother with less pronounced minimum and maximum in comparison with the curve for PVAΔ32. This could be explained by the effect of structural polymorphism on the SAXS curves and/or higher polydispersity of the intact virions [25].
Fig. 3. a) Experimental SAXS curves from intact PVA particles (1) and degraded PVAΔ32 virions (2); b) Kratky plots for intact PVA particles (1) and degraded PVAΔ32 virions (2).
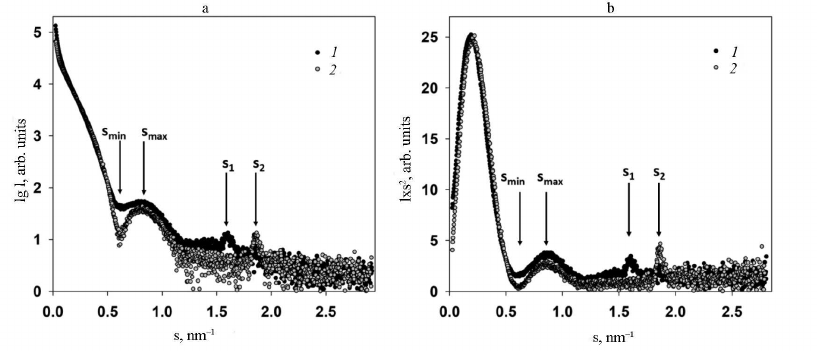
The dissimilarity of the SAXS curves was also observed at the smallest angles in the interval of wave vectors s from 0.08 to 0.25 nm–1: intact virions produced slightly higher scattering intensity, which implies larger amount of scattering matter and indirectly confirms the presence of the N-terminal domain in the CP in the intact virions and its absence in PVAΔ32. Another important difference of the scattering profiles was different position of the Bragg peaks on the SAXS curves from PVA and PVAΔ32 (Fig. 3).
The emergence of the Bragg peaks indicates a certain level of the sample ordering. Considering that the Bragg scattering is characteristic of crystals, the ordered regions are called quasi-crystalline, and their structural characteristics are determined from the equations (1) and (2) (see Materials and Methods). The period of the structures determined from these equations d = 2π/s, the size of the crystallinity regions L, and the degree of disorder Δ/d are presented in the table.
Structural characteristics of intact PVA virions and PVAΔ32
virions
It follows from the Table that the virion with the CP lacking 32 amino acids, PVAΔ32, is significantly more structured: it has larger size of the structured regions L and lesser degree of disorder Δ/d. It is also important for the structural analysis that the period d, which is the distance between the repeating motives of the structure, decreased by 0.5 nm as a result of deletion of 32 amino acid residues from the CP N-terminal domain. These changes refer to small, intermolecular distances and represented alterations within the protein coat that cannot be interpreted as interactions between the virions such as, for example, formation of ordered clusters of these large particles. In the latter case, the period d could not be less than the virion diameter even at the densest packing. Moreover, neither the concentration dependencies, nor the emergence of the structural peak at the smallest angles were observed during SAXS recording, which confirmed the absence of interparticle interactions and cluster formation [26].
The fact that deletion of the N-terminal domain in CP did not cause virion destruction and that its general shape remained preserved with the protein coat internal rearrangement is corroborated by the SAXS curves in Kratky coordinates (Fig. 3b). These graphs demonstrate almost complete match of the scattering profiles and are bell-shaped, which indicates compactness and high order of the structure [12]; but, at the same time, the shift of Bragg peaks on these curves indicates differences in the quasi-crystalline structure, i.e., changes at the molecular level.
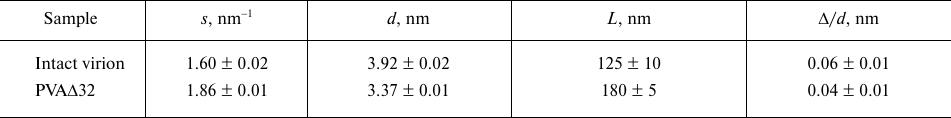
The shapes of the PVA and PVAΔ32 virions at a resolution of 1-2 nm from scattering curves (1 and 5, Fig. 4a) can be elucidated using the ab initio protocol and the DAMMIN program [21]. The distance distribution functions p(r) constructed for this purpose are presented in Fig. 4b.
Fig. 4. a) Experimental scattering curves (1 and 5); curves calculated with the GNOM program based on the distance distribution functions p(r) (2 and 6); scattering curves from the shapes reconstructed ab initio with the DAMMIN program (3 and 7); scattering curves reconstructed ab initio with the MONSA program (4 and 8) for PVA and PVAΔ32, respectively. The groups of the scattering curves for PVA and PVAΔ32 are shifted vertically relative to each other for better visualization; b) distance distribution functions from PVA (1) and PVAΔ32 (2). Inset: distance distribution functions p(r) in the cross-section analysis mode for PVA (1) and PVAΔ32 (2); c) shapes of PVA (1) and PVAΔ32 (2) virions reconstructed with the DAMMIN program; d) shapes of PVA (1) and PVAΔ32 (2) virions reconstructed with the MONSA program. Protein coat is presented as grey virtual atoms (beads), viral RNA – as red beads.
Taking into account the fact that the PVA virions are long flexible filaments, the distance distribution functions p(r) were calculated, firstly, for the ab initio determination of the virion shape as a whole and, secondly, for analysis of its cross-section size. The latter is necessary because of possible changes in the filament diameter following deletion of 32 amino acids from the CP N-terminal region, which is presumably located on the outer surface of the virion. The mode for the analysis of the cross-section of an elongated particle is available as one of the options of the GNOM program [20].
As one can see in Fig. 4b, the distance distribution function p(r) reflecting the general shape of the virions is typical for elongated scattering objects with a cross-section of about 10-16 nm [27]. More accurate analysis of the cross-section size distribution (Fig. 4b, inset) shows that the maximal size of the cross section in the intact virion could reach up to 18 nm, but does not exceed 16 nm for PVAΔ32. The maximum of p(r) for PVA is located at R = 6 nm, thus making the diameter of the intact virion 12 nm, while R = 5.4 nm for the PVAΔ32 indicates that that the virion diameter indeed becomes slightly smaller after removal of 32 N-terminal amino acids.
The low-resolution structures of PVA and PVAΔ32 reconstructed with the DAMMIN program are presented in Fig. 4c, 1 and 2, respectively. Based on the fact that the virions are shaped as long cylinders (filaments), the search for the solution was conducted in a cylindrical region with the length Dmax = 105 nm (Fig. 4b, main panel) and diameters obtained in the analysis of the cross-sections of the virion filaments (Fig. 4b, inset). The scattering curves from the models (Fig. 4a, curves 3 and 7) demonstrate good agreement with the experimental data with χ2 = 2.3 for PVA and χ2 = 1.8 for PVAΔ32. Certain periodicity is noticeable in the PVAΔ32 structure, while the intact virion is less ordered and its general shape is significantly affected by the flexibility of the filaments.
The ab initio virus model was produced using a more universal approach that involved simultaneous reconstruction of both the virion general shape and structures of its individual components – protein and RNA. The two-phase model with separate protein and RNA parts was constructed with the MONSA program [21]. Two cylinders were used as search volumes. The first one used for RNA modeling represented an inner solid cylinder with a 3.2-nm radius and 105-nm length both for PVA and partially degraded virion. The second cylinder was used as a search volume for the protein coat; it was hollow and located outside of the first cylinder. The outer radius of this cylinder was 8.0 nm for the intact virion and 7.0 nm for PVAΔ32, in accordance with the results of the cross-section evaluation. The inner radius of the second cylinder was 3.5 nm for both virions. In the process of modeling, virtual atoms (beads) in the outer cylinder could be assigned phase index 1 or 0 (i.e., protein or solvent), and in the inner cylinder – 2 or 0 (i.e., RNA or solvent). In this way, an adequacy of the model was addressed with CP being on the outer surface and RNA being inside. The reconstructed two-phase models presented in Fig. 4d are approximated with distorted cylinders of the respective sizes and are in good agreement with the models reconstructed with the DAMMIN program. Distortions (imperfections) of the obtained shapes indicate both the effect of the virion flexibility and partial disordering of the structure as a whole and CPs in particular. Interestingly, the beads modeling RNA are spread along the entire axis of the cylinder, which is in agreement with the common concepts of the virus structure. The two-phase models approximate well the experimental SAXS curves (Fig. 4a, curves 4 and 8) with χ2 = 3.4. It must be mentioned that the Bragg peaks in the regions s = 1.6 and 1.9 nm–1 are also well reproduced. The presence of Bragg peaks reflects regular helical CP packing, which is observed in the produced models with the structure periodicity more pronounced for the PVAΔ32.
Considering that the inverse problem in SAXS, which is determination of the three-dimensional structure of an object from the one-dimensional scattering curve, is generally ambiguous, it is necessary to estimate the corridor width for the possible solutions. This is carried out with the AMBIMETER program [19]. This tool allows fast evaluation of the degree of ambiguity of an arbitrary scattering profile from the monodisperse solution of the uniform particles and is based on the vast library of scattering diagrams from the shape patterns defining the variety of topologies of the low-resolution structures. The number of patterns similar to the experimental SAXS data provide the degree of ambiguity associated with the data, and the logarithm of this value is used in the AMBIMETER program as a quantitative parameter. As a rule, the value below 1.5 suggests that the solution is unique, and estimation above 2.5 indicates ambiguous shape reconstruction.
In this study, only one compatible shape was found for PVAΔ32, which indicated narrow range of similar shapes of the low-resolution structures. In the case of intact PVA, 88 shape patterns were found to be compatible with the data, and the respective estimate of ambiguity was 1.9, which indicated possible uncertainty of the shape reconstruction for this sample due to the flexibility of the intact virions.
In general, the ab initio reconstruction of the virion shape from the SAXS data demonstrated noticeable changes in the virion structure after deletion of 32 amino acids from CP N-terminal domain. These changes involve both the size of the virion filament cross-section and the periodicity of the CP structure. The decrease in the period d value is presumably associated with the pitch reduction of the helical protein packing in the virus coat.
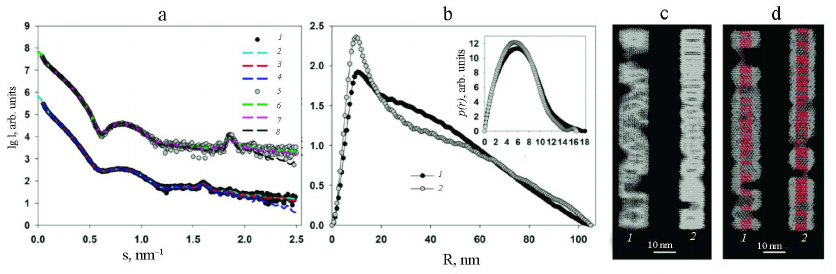
In order to show the correctness of this assumption, the structure of PVA and PVAΔ32 was modeled with geometric bodies. For this purpose, bead models of helical shapes were constructed with the external dimensions determined from the analysis of the distance distribution functions p(r). The small-angel scattering curves from the obtained helices were calculated and compared with the experimental SAXS curves from PVA and PVAΔ32 (Fig. 5a).
Fig. 5. Modeling of the structures of intact and partially degraded virions. a) Experimental SAXS curves (1 and 3) and scattering from the helical shapes (2 and 4) for PVA and PVAΔ32, respectively. Modeled Bragg peaks are indicated with arrows. b) Helical models imitating intact virion (1) and PVAΔ32 (2).
Modeling with geometric bodies is a technique of rough approximation, which cannot account for all features of the real structures, their mobility, and flexibility of individual fragment, but allows constructing models of selected elements important for analysis. In our case, we needed to prove that the Bragg peaks on the experimental scattering curves represent the pitch of the helical protein packing in the virus coat. For this purpose, the distance between the turns of the model helices was set in accordance with the characteristic period d =2π/s, which was d = 3.9 nm for the intact virion and d = 3.4 nm for the partially degraded virion (Table). The width of the helical filaments was selected equal to 3.4 nm based on the suggestion that this value cannot be larger than the helix pitch for the PVAΔ32. Slight changes toward reduction did not affect the results of modeling. As can be seen in Fig. 5a, the maxima of the scattering intensity from the model structure adequately describe Bragg peaks on the experimental curves. Hence, it can be concluded that deletion of 32 amino acid residues from the N-terminal domain of PVA CP indeed causes changes in the helical pitch of the protein coat – the helix contracts and the virion become more rigid and, hence, is better approximated by the cylindrical shape (Fig. 4).
It could be suggested based on the obtained results that the N-terminal domain is partially located between the turns of the packed CP, facilitating interaction with neighboring protein subunits and keeping the helical turns at a certain distance. On the other hand, a part of the N-terminal domain protrudes from the virion surface, which is manifested by a larger diameter of the intact virion in comparison with the partly degraded one. It is important to mention that the unstructured regions of protein molecules serve as binding elements in the interaction with various proteins and other cell components [5]. At the same time, the cell-to-cell transport of vital metabolites occurs through the plasmodesmata; despite their large sizes, plant pathogens can also penetrate into cells through the plasmodesmata. For this purpose, viruses selectively use, for example, some cellular myosins [28]. Hence, it could be suggested that the disordered flexible fragments of the CP N-terminal domain in PVA could serve as mediators for the binding with certain cellular structures to facilitate the cell-to-cell virus transport. In this case, direct removal of these virion fragments with respective agents could result in the pathogen deactivation.
Virion flexibility is another parameter that is very important for the virus penetration into the cell. It is known that flexibility increases with the increase of the virion length in the following series: tobamoviruses (300 nm), potexviruses (500-1000 nm), closterovirused (1400-2000 nm), which facilitates their intercellular transport through the plasmodesmata. That means that the longer is the virus the more flexible it should be to penetrate into the cell. It is likely that it is exactly the CP N-terminal domain that defines the flexibility of virions. The data obtained in this study corroborate this suggestion.
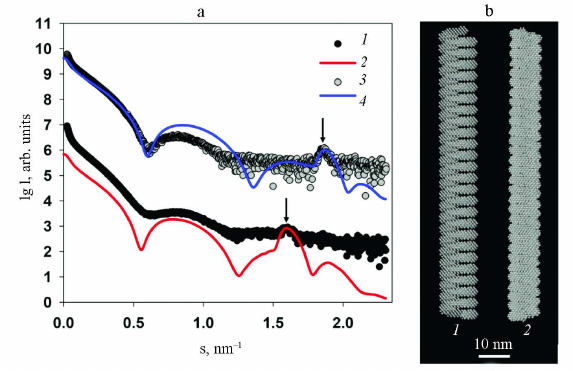
The high-resolution structure of the PVA has not been obtained yet; however, recently, the structure of another potyvirus, PVY, whose CP displays a very high homology with the PVA CP, was solved with a 3.4 Å resolution using cryoelectron microscopy [3]. Therefore, we attempted to compare the high-resolution structure of PVY with the structure of PVA obtained by SAXS and the method of rigid body modeling. The primary structures of CPs from PVA and PVY are very similar, except the N-terminal residues. It was determined using pairwise alignment of amino acid sequences by the LALIGN program that the CPs from these two viruses contain 64% identical and 91% similar residues among 237 residues (without 32 N-terminal residues) [24] (Fig. 6).
Fig. 6. Alignment of the amino acid sequences of PVA and PVY CPs with the LALIGN program (Expasy). Identical residues are marked with asterisks (*); semiconserved replacements are shown with dots (·). The sequences deleted with trypsin (for PVAΔ32) or absent in PDB ID: 6HXX (for PVY) are underlined. The structures of PVA CP no. GI:25013598 and PVY CP (strain NTN) no. GI:728041063 are presented.

Because of the high identity of the amino acid sequences of the CPs from these two viruses, it was possible to use the 3.4 Å-resolution structure of the PVY CP (PDB: 6HXX) for modeling the structure of protein coat of PVA. The packing of PVY CP is characterized by the helical symmetry; the fragment used for modeling consisted of 35 protein subunits including atomic coordinates of amino acid residues 44 to 267, i.e., without 43 N-terminal residues, for which atomic coordinates have not been determined because of the low electron density of these unstructured fragments. The PVY fragment was repeatedly reproduced along the helical axis using the MASSHA program [22] to construct a model containing 30 turns (260 subunits) with the maximum length of 105 nm. The number of turns was selected to obtained the best possible agreement with the experimental data and scattering curves. Comparison of the experimental and model scattering profiles demonstrated a certain deviation of the curves in the 0.08 < s < 1.2 nm–1 range, as well as a slight shift of the Bragg peak with the maximum at s = 1.9 nm–1 (d = 3.4 nm) towards larger sizes with s = 1.8 nm–1 (d = 3.5 nm). These deviations indicate differences in the structure of the protein coats of the PVA and PVY particles despite the fact that they are highly homologous. In addition, one must take into account that the obtained model is closer not to the intact PVA virion, but to its partly degraded form. Moreover, the deviations between the experimental and model curves with χ2 ~ 25 (Fig. 7) reflect the imperfectness of the virion real structure in comparison with its solid model on the atomic level, which cannot change its conformation. At the same time, biological measurements of the SAXS are made in solution, where the scattering particles preserve their main characteristics including flexibility.
Fig. 7. Results of modeling of the PVA protein coat using high-resolution structural blocks of the PVY CP (PDB: 6HXX). a) Curves: 1) experimental SAXS curve from PVAΔ32; 2) scattering profile from the constructed model calculated with the CRYSOL program. b) Structural model of the protein coat imitating PVA.
Nevertheless, the obtained high-resolution model of PVA demonstrates undoubted structural similarity of the protein coat in these two potyviruses.
The main conclusion from this part of study is that trypsinization and partial degradation of PVA changes the pitch of the protein coat helical packing. Deletion of 32 amino acids causes decrease in the distance between the helix turns from 3.9 nm to 3.4 nm. Such compression of the helix inevitably produces changes in the flexibility of the partially degraded virion, as a compressed spring is always more rigid that a free helix. This conclusion was confirmed by the transmission electron microscopy.
Electron microscopy examination of PVA particles. According to the TEM data, the intact PVA virions are typical filamentous particles (Fig. 8a), while the trypsinized PVAΔ32 virions are mainly represented by the rod-shaped particles with a smaller diameter (Fig. 8b) that resembled dry spaghetti. The diameter of viral particles was measured using the Gatan program (DigitalMicrograph software) and was found to be 14.4 ± 0.2 nm and 12.1 ± 0.3 nm for the intact and degraded virions, respectively, which also indirectly indicated the removal of the CP N-terminal fragments from the surface of PVA virions.
Fig. 8. Transmission electron microscopy images of intact PVA virions (a) and degraded PVAΔ32 virions (b).

In general, the TEM data are in good agreement with the SAXS results, taking into account different states of the virus during the SAXS measurements (in solution, which is closer to natural conditions) and TEM examination (dried on a support).
In conclusion, for the first time, the low-resolution structure of the filamentous plant virus PVA, both in its intact and partially degraded form, was obtained under conditions close to the natural ones. The results of the SAXS measurements revealed that deletion of 32 N-terminal amino acid residues of CP resulted in the change of the pitch of the protein coat helical packing. We also found that the trypsinized virions exhibited more ordered structure and formed predominantly rod-shaped particles with a diameter smaller than the diameter of flexible particles of the intact virus, which was in good agreement with the TEM results. In general, the conducted structural analysis indicates the significance of the CP N-terminal domain for the PVA vital functions, which should be considered for further development of strategies to fight these plant pathogens.
Funding. This work was supported by the Russian Foundation for Basic Research (project no. 18-04-00525a), by the Ministry of Science and Higher Education of the Russian Federation (agreement 075-15-2019-1653), and by the Ministry of Science and Higher Education of the Russian Federation within the State assignment FSRC “Crystallography and Photonics” RAS (SAXS experiments).
Ethics declarations. The authors declare no conflict of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
REFERENCES
1.Ksenofontov, A. L., Paalme, V., Arutyunyan, A. M.,
Semenyuk, P. I., Fedorova, N. V., et al. (2013) Partially disordered
structure in intravirus coat protein of potyvirus potato virus A,
PLoS One, 8, e67830, doi:
10.1371/journal.pone.0067830.
2.Zamora, M., Mendez-Lopez, E., Agirrezabala, X.,
Cuesta, R., Lavin, J. L., et al. (2017) Potyvirus virion structure
shows conserved protein fold and RNA binding site in ssRNA viruses,
Sci. Adv., 3, eaao2182, doi: 10.1126/sciadv.aao2182.
3.Kezar, A., Kavcic, L., Polak, M., Novacek, J.,
Gutierrez-Aguirre, I., et al. (2019) Structural basis for the
multitasking nature of the potato virus Y coat protein, Sci.
Adv., 5, eaaw3808, doi: 10.1126/sciadv.aaw3808.
4.Cuesta, R., Yuste-Calvo, C., Gil-Carton, D.,
Sanchez, F., Ponz, F., and Valle, M. (2019) Structure of Turnip mosaic
virus and its viral-like particles, Sci. Rep., 9, 15396,
doi: 10.1038/s41598-019-51823-4.
5.Uversky, V. N. (2013) Unusual biophysics of
intrinsically disordered proteins, Biochim. Biophys. Acta,
1834, 932-951, doi: 10.1016/j.bbapap.2012.12.008.
6.Ksenofontov, A. L., Parshina, E. Y., Fedorova, N.
V., Arutyunyan, A. M., Rumvolt, R., et al. (2016) Heating-induced
transition of Potyvirus Potato Virus A coat protein into
beta-structure, J. Biomol. Struct. Dyn., 34, 250-258,
doi: 10.1080/07391102.2015.1022604.
7.Ksenofontov, A. L., Dobrov, E. N., Fedorova, N. V.,
Arutyunyan, A. M., et al. (2018) Structure of Potato Virus A coat
protein particles and their dissociation, Mol. Biol. (Mosk.),
52, 1055-1065, doi: 10.1134/S0026898418060101.
8.Ksenofontov, A. L., Dobrov, E. N., Fedorova, N. V.,
Serebryakova, M. V., Prusov, A. N., et al. (2018) Isolated Potato Virus
A coat protein possesses unusual properties and forms different short
virus-like particles, J. Biomol. Struct. Dyn., 36, 1-11,
doi: 10.1080/07391102.2017.1333457.
9.Anindya, R., and Savithri, H. S. (2003)
Surface-exposed amino- and carboxy-terminal residues are crucial for
the initiation of assembly in Pepper vein banding virus: a flexuous
rod-shaped virus, Virology, 316, 325-336, doi:
10.1016/s0042-6822(03)00593-2.
10.Tatineni, S., McMechan, A. J., and Hein, G. L.
(2018) Wheat streak mosaic virus coat protein is a determinant for
vector transmission by the wheat curl mite, Virology,
514, 42-49, doi: 10.1016/j.virol.2017.10.018.
11.Voloudakis, A. E., Malpica, C. A.,
Aleman-Verdaguer, M. E., Stark, D. M., Fauquet, C. M., and Beachy, R.
N. (2004) Structural characterization of Tobacco etch virus coat
protein mutants, Arch. Virol., 149, 699-712, doi:
10.1007/s00705-003-0247-x.
12.Svergun, D. I., Koch, M. H. J., Timmins, P. A.,
and May, R. P. (2013) Small Angle X-Ray and Neutron Scattering from
Solutions of Biological Macromolecules, First Edn., Oxford
University Press, Oxford, doi:
10.1093/acprof:oso/9780199639533.001.0001.
13.Franke, D., Petoukhov, M. V., Konarev, P. V.,
Panjkovich, A., Tuukkanen, A., et al. (2017) ATSAS 2.8: a comprehensive
data analysis suite for small-angle scattering from macromolecular
solutions, J. Appl. Crystallogr., 50, 1212-1225, doi:
10.1107/S1600576717007786.
14.Ksenofontov, A. L., Petoukhov, M. V., Prusov, A.
N., Fedorova, N. V., and Shtykova, E. V. (2020) Characterization of
tobacco mosaic virus virions and repolymerized coat protein aggregates
in solution by small-angle x-ray scattering, Biochemistry
(Moscow), 85, 310-317, doi: 10.1134/s0006297920030062.
15.Laemmli, U. K. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685, doi: 10.1038/227680a0.
16.Ksenofontov, A. L., Kozlovskii, V. S.,
Kordiukova, L. V., Radiukhin, V. A., Timofeeva, A. V., and Dobrov, E.
N. (2006) Determination of concentration and aggregate size in
influenza virus preparations using the true UV-absorption spectra,
Mol. Biol., 40, 152-158, doi:
10.1134/S0026893306010201.
17.Blanchet, C. E., Spilotros, A., Schwemmer, F.,
Graewert, M. A., Kikhney, A., et al. (2015) Versatile sample
environments and automation for biological solution X-ray scattering
experiments at the P12 beamline (PETRA III, DESY), J. Appl.
Crystallogr., 48, 431-443, doi:
10.1107/S160057671500254X.
18.Konarev, P. V., Volkov, V. V., Sokolova, A. V.,
Koch, M. H. J., and Svergun, D. I. (2003) PRIMUS: a Windows PC-based
system for small-angle scattering data analysis, J. Appl.
Crystallogr., 36, 1277-1282, doi:
10.1107/s0021889803012779.
19.Petoukhov, M. V., and Svergun, D. I. (2015)
Ambiguity assessment of small-angle scattering curves from monodisperse
systems, Acta Crystallogr. D Biol. Crystallogr., 71,
1051-1058, doi: 10.1107/s1399004715002576.
20.Svergun, D. I. (1992) Determination of the
regularization parameter in indirect-transform methods using perceptual
criteria, J. Appl. Crystallogr., 25, 495-503, doi:
10.1107/s0021889892001663.
21.Svergun, D. I. (1999) Restoring low resolution
structure of biological macromolecules from solution scattering using
simulated annealing, Biophys. J., 76, 2879-2886, doi:
10.1016/S0006-3495(99)77443-6.
22.Konarev, P. V., Petoukhov, M. V., and Svergun, D.
I. (2001) MASSHA – a graphics system for rigid-body modelling of
macromolecular complexes against solution scattering data, J. Appl.
Crystallogr., 34, 527-532, doi:
10.1107/s0021889801006100.
23.Svergun, D., Barberato, C., and Koch, M. H. J.
(1995) CRYSOL – a program to evaluate x-ray solution scattering
of biological macromolecules from atomic coordinates, J. Appl.
Crystallogr., 28, 768-773, doi:
10.1107/s0021889895007047.
24.Artimo, P., Jonnalagedda, M., Arnold, K.,
Baratin, D., Csardi, G., et al. (2012) ExPASy: SIB bioinformatics
resource portal, Nucleic Acids Res., 40, W597-603, doi:
10.1093/nar/gks400.
25.Shtykova, E. V. (2015) Shape determination of
polydisperse and polymorphic nanoobjects from small-angle X-ray
scattering data (computer simulation), Nanotechnol. Russia,
10, 408-419, doi: 10.1134/S1995078015030155.
26.Tardieu, A. (1994) Neutron and synchrotron
radiation for condensed matter studies. Applications to Soft Condensed
Matter and Biology, vol III (Les editions de Physique (France))
(Berlin: Springer), pp. 145-160.
27.Feigin, L. A., and Svergun, D. (1987) Structure
analysis by small-angle x-ray and neutron scattering, Plenum Press, New
York, p. 335.
28.Amari, K., Lerich, A., Schmitt-Keichinger, C.,
Dolja, V. V., and Ritzenthaler, C. (2011) Tubule-guided cell-to-cell
movement of a plant virus requires class XI myosin motors, PLoS
Pathog., 7, e1002327, doi: 10.1371/journal.ppat.1002327.