REVIEW: A Code Within a Code: How Codons Fine-Tune Protein Folding in the Cell
Anton A. Komar1,2,3,4*
1Center for Gene Regulation in Health and Disease and Department of Biological, Geological and Environmental Sciences, Cleveland State University, Cleveland, Ohio 44115, USA2Department of Biochemistry and Center for RNA Science and Therapeutics, Case Western Reserve University, Cleveland, Ohio 44106, USA
3Genomic Medicine Institute, Lerner Research Institute, Cleveland Clinic, Cleveland, Ohio 44195, USA
4DAPCEL, Inc., Cleveland, Ohio 44106, USA
Received April 11, 2021; Revised April 11, 2021; Accepted April 28, 2021
The genetic code sets the correspondence between the sequence of a given nucleotide triplet in an mRNA molecule, called a codon, and the amino acid that is added to the growing polypeptide chain during protein synthesis. With four bases (A, G, U, and C), there are 64 possible triplet codons: 61 sense codons (encoding amino acids) and 3 nonsense codons (so-called, stop codons that define termination of translation). In most organisms, there are 20 common/standard amino acids used in protein synthesis; thus, the genetic code is redundant with most amino acids (with the exception of Met and Trp) are being encoded by more than one (synonymous) codon. Synonymous codons were initially presumed to have entirely equivalent functions, however, the finding that synonymous codons are not present at equal frequencies in mRNA suggested that the specific codon choice might have functional implications beyond coding for amino acid. Observation of nonequivalent use of codons in mRNAs implied a possibility of the existence of auxiliary information in the genetic code. Indeed, it has been found that genetic code contains several layers of such additional information and that synonymous codons are strategically placed within mRNAs to ensure a particular translation kinetics facilitating and fine-tuning co-translational protein folding in the cell via step-wise/sequential structuring of distinct regions of the polypeptide chain emerging from the ribosome at different points in time. This review summarizes key findings in the field that have identified the role of synonymous codons and their usage in protein folding in the cell.
KEY WORDS: genetic code, codon usage, synonymous codons, translation kinetics, nascent peptides, co-translational protein foldingDOI: 10.1134/S0006297921080083
Abbreviations: ADAMTS13, a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13; CAT, chloramphenicol acetyltransferase; FRET, fluorescence resonance energy transfer; mRNA, messenger RNA; NMR, Nuclear magnetic resonance; PGK, phosphoglycerate kinase; P-gp, P-glycoprotein; RNase A, Ribonuclease A; tRNA, transport RNA.
INTRODUCTION
Sixty years ago, Crick, Barnett, Brenner, and Watts-Tobin postulated key features of the genetic code [1]. They suggested that a group of three bases (a codon) encodes one amino-acid; that the code is not overlapping, that the sequence of the bases is read from a fixed starting point, that the code does not contain any special “commas” to show how to select the right triplets and that the code is “degenerate”; that is, in general, one particular amino acid can be encoded by one of several (synonymous) codons [1]. Subsequent studies on the responses of aminoacyl-transport RNAs (tRNAs) to trinucleotide templates allowed the general patterns of decoding to be established and to reveal the exact nature of degeneracy of the genetic code [2-8].
It was further recognized, that synonymous codons, while coding for the same amino acid, might not necessarily be entirely equivalent [9-19]. It was found that synonymous codons are decoded with different rates/efficiencies (and stringencies) and that the frequencies of occurrence of the synonymous codons largely determine the rate at which the synonymous codons are decoded [20-22]. It should be noted in this regard that the rate/efficiency at which a codon is translated is largely determined by the availability of cognate tRNA(s), whereas fidelity is generally impacted by the competition with noncognate tRNA(s) [23-27]. As it was found that the codon frequencies usually correlate with the availability/concentration of cognate tRNAs [12, 13, 16], it was suggested that the codon choice affects translation elongation rates [20-22] (tRNA concentration appeared to be one of the major determinants affecting decoding times [28, 29]). Indeed, many subsequent experiments showed that the frequently used codons are translated more rapidly than the infrequently used ones due to the higher availability (during decoding of the message) of the corresponding frequent cognate tRNAs [20-22, 30, 31]. In line with these observations, the highly expressed genes were also found to harbor more preferred/frequent codons compared to the lowly expressed genes, which were found to be enriched in synonymous un-preferred codons [17, 18]. And while optimal/frequent codons were found to differ among species, coordinated with changes in the population of tRNA genes [13, 16], it was concluded that the messenger RNA (mRNA) encodes not only the amino acid sequence of the protein, but also specifies the protein expression levels [32-36]. A theory of codon-tRNA co-evolution (necessary to balance accurate and efficient protein production) was put forward [32-36]. It has been proposed that selection favors optimal/frequent codons over un-preferred/rare codons to support efficient protein production [32-36]. However, it has later became clear that the un-preferred codons are also subjects to constraints and have important functional roles, particularly in mRNA homeostasis/intracellular stability [37-44] and protein folding in the cell (see below).
In parallel with the efforts to establish the nature of the genetic code and the decoding process, it was determined that the amino acid sequence of a protein contains the protein folding code, i.e., information necessary to specify its unique three-dimensional structure [45]. While the exact nature of the protein folding code was (and in part remains) unknown, it was further concluded that the native folded state of a protein is not achieved by a random search among all possible conformations en route to the native structure (which would require enormous amount of time to accomplish), but likely is achieved through a set of limited and well-defined pathways exploring just a set of available conformational spaces (see reviews [46-51]).
Almost at the same time (in early 1960s and 1970s), it was also determined that in vivo protein folding, at least for some proteins, is a co-translational process (i.e., it occurs during protein synthesis) [52-58]. What was unclear then, whether the ribosome and/or the process of translation can impact and/or fine-tune the process of protein folding and to what extent a specific pathway of protein folding in the cell (if it exists) may ensure high efficiency of the in vivo protein folding. The answers to these questions slowly emerged in the last 5 decades and continue to emerge now.
Decoding, or translation, of mRNAs is performed by ribosomes, with addition of each new amino acid to the growing polypeptide chain. It can thus be hypothesized that the nature of the decoding process, the ribosome (as a large macromolecular complex), and/or the rate of polypeptide polymerization may affect the final conformation of the protein, by influencing conformational space of the growing polypeptide chain and/or by changing the kinetics of protein folding during polypeptide synthesis, or both. Many recent experiments indicated that it is indeed the case and that the mechanism of decoding, the ribosome itself, and, importantly, the kinetics of translation, which defines the rate of polypeptide chain polymerization may affect the process and efficiency of the in vivo protein folding (see below). However, it was not until 1985, when stereochemical analysis of the ribosomal transpeptidation reaction performed by Lim and Spirin, allowed to suggest that the decoding process can impact protein folding and that the ribosome may facilitate generation of an alpha-helical conformation in the growing nascent chain [59, 60], therefore supporting the view that translation is not merely a process of sequential addition of amino acids to the growing polypeptide, but a process that may also influence the mechanism of protein folding.
Subsequently, we and others showed that variations in the local translation rates primarily governed by nonuniform and non-random utilization of the synonymous codons along mRNA, may affect/facilitate protein folding by allowing ordered, sequential structuring of the discrete nascent polypeptide chain portions synthesized by the ribosome and that kinetics of protein synthesis may serve as a secondary code fine-tuning protein folding in the cell (see review [61]). Moreover, analyses of the codon distribution along mRNA(s) allowed us and others to find “imprints” of the protein structure embedded in mRNA sequences [61]. It became clear that selection not only supports efficient protein expression, but also favors a specific non-random distribution of codons along the message and that mutational pressure and genetic drift position the un-preferred codons at specific locations to have important functional role(s) and, specifically, in protein folding [61].
Below, I’ll review some of the key findings supporting existence of the secondary code within the genetic code that helps fine-tune protein folding in the cell and explaining the role of rare/un-preferred codons in this process.
THE CODE FOR PROTEIN FOLDING
Seminal experiments performed by Christian Anfinsen and his colleagues (in the 1950-60s) on the reversible denaturation of ribonuclease [45] and many similar subsequent experiments by other researchers, which used a multitude of different other proteins indicated that amino acid sequence of a protein contains all the information necessary to specify its unique three-dimensional structure [46-51]. Many theoretical and computational studies, including recent breakthrough research showing that it is possible to accurately predict protein structures from the amino acid sequence [62, 63] further supported this key postulate of the “protein folding problem”. In some (most recent) cases, in silico structure predictions appeared to be indistinguishable from the structures determined using X-ray crystallography [64], which is a considerable advance in the field and an important proof of the Anfinsen’s postulate.
The concept of the folding funnel suggesting that there could be distinct multiple pathways, which guide protein folding to a native conformation with the lowest free energy minimum, has been developed to explain how proteins fold (see reviews [46-51]). This concept was further amended by the new model for protein folding, which described the folding process through formation of the separately cooperative folding units (foldons) (see reviews [65-67]). Several proteins have been shown to fold via a sequential pathway consisting of such foldon-based intermediates [65-67]. The inherently cooperative nature of foldons was suggested to determine rapid formation of the native structure [65-67]. The foldon hypothesis was also able to satisfy the requirement for the energetic bias toward native-like interactions. The limited number and the small size of such foldons (~15-35 residues) provided an additional solution to the Levinthal’s time-scale paradox problem [68].
For many (especially small) proteins, theoretical studies, computational and in vitro unfolding/refolding experiments have matched up (see [69] and ref. therein). These experiments provided general answers to some of the key questions in the field, namely: how do proteins fold, and why do they fold in that way?
However, in many cases (and especially in the cases of large proteins) these experiments could not describe the process in full [70]. In addition, many attempts to achieve refolding of the isolated denatured proteins in a test tube were also only partially successful. Importantly, refolding in a test tube was found to be exceedingly slow [46] and for some proteins could take hours or even days [71, 72]. At the same time, it has been well known that this folding rate “incompatibility problem” can be easily alleviated in vivo. It thus became clear that the co-translational nature of the protein folding process in the cell may be a key to understanding of the natural code for protein folding. Researchers working on the protein folding theory, and employing computational and bench experiments have turned their attention to the in vivo process aiming to understand the similarities and differences between the in vitro and in vivo protein folding pathways and mechanisms (see reviews [73-78]).
It must be noted in this regard that the co-translational protein folding is a process that is substantially different from the refolding in vitro [79-84]. It is the process that is characterized by the attachment of the nascent polypeptide chain (through its C-terminal end) to the ribosome during protein synthesis, which reduces conformational space and degrees of freedom of the growing chain, therefore significantly limiting the number of possible intermediates and thus also reducing the number of possible folding pathways [79-84]. It must be also noted that the ribosome serves as a hub [85] for many molecular chaperones and itself was found to possess intrinsic “chaperone-like” protein folding activity (see [86-88] and ref. therein). Molecular chaperones and folding catalysts might interact with the growing nascent chain emerging from the ribosomal tunnel, thereby accelerating the slow steps in protein folding and preventing misfolding [73-78]. However, it has been suggested that both chaperones and folding catalysts are involved mainly in kinetic partitioning between the proper folding and aggregation; thus, they are thought to affect the yield of the correctly folded protein rather than the folding mechanism [73-78]. Co-translational protein folding is a process that begins very early during the process of polypeptide chain synthesis on the ribosome, with some secondary structure elements (such as, e.g., alpha-helices) forming inside the ribosomal tunnel [89-92] and some tertiary structures forming inside the vestibule (lower/wider) region of the ribosomal exit tunnel [93-96] (Fig. 1). Importantly, co-translational folding is a “vectorial” process, whereby folding of the nascent polypeptide proceeds from its N- to C-terminal end, and results in a sequential structuring of the distinct regions of the polypeptide emerging from the ribosomal tunnel [61, 79-84].
Fig. 1. Co-translational folding is a stepwise vectorial process proceeding in most of the cases from the N- to C-terminal end of the growing polypeptide chain. The appearance of distinct folding intermediates is linked to the overall and local rates of translation. The process begins very early during polypeptide chain synthesis on the ribosome, with some secondary structure elements (such as, e.g., alpha-helices) forming inside the ribosomal tunnel (and some tertiary structures forming inside the vestibule (lower/wider) region of the ribosomal exit tunnel. The tertiary structure of the protein is nearly formed at the end of the protein synthesis.
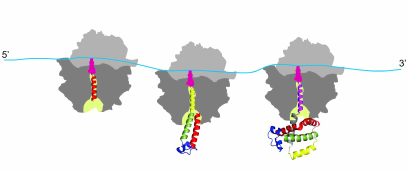
While exceptions to this general rule have been found, where it was shown that synthesis of the polypeptide on the ribosome can run counter to the directional folding of a nascent protein domain (see [97] and ref. therein), it was nevertheless concluded that for the majority of proteins the synthesis and co-translational folding proceed concomitantly [61, 79]. Finally, co-translational protein folding is tightly linked to translation elongation, which is not a uniform process (see [61, 79, 98] and ref. therein). Protein synthesis rates were suggested to be evolutionarily tuned to optimize protein folding, allowing proteins to circumvent deep kinetic traps during the co-translational folding (see [61, 79, 98-107] and ref. therein). It was therefore postulated that in vivo protein self-assembly is coordinated with protein synthesis and evolutionary optimized to fast yield correctly folded proteins with high efficiency (see [61, 79, 98-107] and ref. therein). We and others suggested (see below) that the rare codons (that are translated slowly) may be under evolutionary selection to allow for efficient co-translational folding and that translation pause sites may define the window of opportunity for the protein parts to fold locally, particularly at the critical points where folding is far from equilibrium (see [61, 79, 98-107] and ref. therein). It must be noted however, that many details of the co-translational folding process remain only partially understood and both the mechanism and the pathway of co-translational folding remain subjects of intense investigation these days.
CO-TRANSLATIONAL PROTEIN FOLDING: PROGRESS IN
UNDERSTANDING
The progress that has been achieved in understanding the co-translational protein folding has been tightly linked to the development of novel methods and approaches (see review [82]).
The studies of co-translational protein folding can roughly be subdivided into three major periods. In the early 1960s and 1970s, the first observations were made suggesting that in vivo protein folding, at least for some proteins, is a co-translational process [52-58]. These were mainly steady-state experiments that established a basic set of requirements for methods aimed at studying co-translational folding, such as that (i) there should be an easily measurable means for assessment of proper folding of nascent chains on the ribosome; (ii) it must be ensured that the specific structural features under investigation are indeed attributable to the ribosome-bound nascent chains and not to the polypeptide chains bound to ribosomes/polyribosomes nonspecifically; (iii) the polypeptide chains should be synthesized de novo to ensure that the measured outcomes are truly the result of a co-translational process. Majority of these early experiments involved isolation/fractionation (from cellular extracts) of the ribosome-bound nascent chain complexes through a sucrose density gradient, followed by the assessment of structural properties of the nascent chains through measurement of (i) their specific enzymatic activities [52-54], (ii) their recognition by specific/conformational antibodies [55], or (iii) formation of correct disulfide cross-bridges within and/or between the nascent chains [56-58]. These initial experiments demonstrated that co-translational folding does take place, however understanding of the mechanism and the extent of this phenomenon were limited then.
The research on co-translational folding then languished due to domination of the in vitro denaturation/renaturation studies. Renewed interest has been generated in late 1980s and mid 1990s with recognition that the process may not be limited to just a few isolated cases, but may be characteristic to many proteins in the cell (see review [82]). The research in this area has been also facilitated by massive application of the cell-free in vitro translation systems (such as, e.g., Rabbit Reticulocyte Lysate (RRL) system developed by Pelham and Jackson in 1976 [108]), introduction of new model proteins (and, in particular, chemiluminescent and fluorescent proteins), and development of new kinetic approaches that paved the way to studies of co-translational protein folding in real time.
In this regard, the study by Kolb, Makeyev, and Spirin is of special interest as it was one of the first attempts to investigate co-translational protein folding in real-time. The authors developed a technique to continuously monitor enzymatic activity of a newly synthesized firefly luciferase in a cell-free system in a luminometer cuvette [109]. Luciferase activity indicative of folding of the protein was detected as soon as the full-length molecule was formed in the translation reaction [109]. Importantly, addition of RNase A abrogated both translation and accumulation of the active luciferase, indicating that synthesis and folding of firefly luciferase proceed concomitantly. While the authors were unable to detect any luciferase activity in the ribosome-bound chains (the last 12 C-terminal amino acids were found to be important for activity of the enzyme ([109] and ref. therein), they nevertheless found that firefly luciferase became active immediately after the release of the nascent chains [109]. Importantly, such rapid acquisition of the enzyme activity was incompatible with the post-translational folding scenario, as refolding of the enzyme from the denatured state was much slower [109].
At the same time, our group at the Department of Molecular Biology at Moscow State University together with Alexander S. Spirin’s laboratory at the Institute of Protein Research in Pushchino used heme binding to probe co-translational folding of the alpha-globin chains [110, 111]. The ability of co-factors and ligands (such as heme) to specifically bind to growing polypeptide chains has been viewed as an indication that a binding-competent conformation has been achieved on the ribosome [82, 110, 111]. Using in vitro translation reactions performed in the presence of [3H]hemin and [14C]leucine or [35S]methionine together with sucrose gradient centrifugation and puromycin treatment, we showed that the ribosome-bound globin chains were capable of efficient heme binding [110, 111]. In addition, we found that the incomplete alpha-globin nascent chains attached to the ribosome are capable of co-translational heme binding, indicating that the structure that allows for heme binding in the nascent chain is achieved prior to the completion of alpha-globin synthesis [111]. Many additional studies in this period of time provided overwhelming support for co-translational folding (see reviews [61, 82]).
The start of the third period in co-translational folding studies can be roughly attributed to the early 2000s. This period is continued now and is characterized by the explosion of techniques and methods to study co-translational folding (such as single-molecule and time-resolved fluorescent approaches [96], allowing to study protein folding in real time [96] and at a single molecule level, applications of nuclear magnetic resonance spectroscopy and cryo-electron microscopy, allowing to understand the structure of ribosome-bound nascent chains at atomic resolution), the development of many other approaches combining in vivo and in vitro (cell-free translation) experiments, as well as substantial advancement of the computational studies allowing to simulate and understand the process of co-translational folding (see review [82]).
It became evident that the co-translational folding is characteristic to almost every protein in the cell of pro- and eukaryotic origin that are single and multidomain, single and multisubunit, cytosolic, secretory and membrane and that without elucidation of its mechanism and pathway comprehensive understanding of the protein folding code would be not possible.
CODON USAGE CODE FOR PROTEIN FOLDING IN THE CELL: THE
NATURE
Investigation of the co-translational protein folding and, specifically, experiments revealing that refolding of proteins from the denatured state can be much slower than co-translational folding, even when compared in the same environment (like, e.g., cellular extracts used for in vitro translation) [109], prompted the suggestion that vectorial nature of the co-translational folding and perhaps additional features present in mRNA beyond the amino acid sequence, might explain efficiency of the in vivo process [112-115]. It became clear however that directionality of the co-translational folding might likely play only a fine-tuning role (yet, very important, as would be seen from the discussion below) in the overall folding process as some proteins, e.g., circularly permuted proteins and proteins obtained during chemical (Merrifield) synthesis, appear to be correctly folded following reversal of the synthesis direction [46, 116].
The studies by Lim and Spirin [59, 60], mentioned above, suggested that decoding on the ribosome may facilitate generation of an alpha-helical conformation in the growing nascent chain. Several questions immediately arose with this assumption: (i) If ribosomes facilitate generation of an alpha-helical conformation, how then other secondary structures (other types of helices, turns, beta-structures, etc.) are formed during the protein synthesis? (ii) Alpha-helices are known to fold fast [46], would then formation of the fast folding units require faster translation rates and formation of the slow folding units correspondingly require slower translation rates? (iii) Could then a time window be necessary for interconversion of an alpha-helix to other secondary structure(s) during protein synthesis on the ribosome? (iv) Could then translation pause sites separate formation of the different structural elements (like secondary structures, of the same and/or different type) during protein synthesis on the ribosome? (v) Would also sequential formation of the larger folding units (like domains in multidomain proteins) require a more extended pause in translation? (vi) Finally, how then changes in the translation kinetics would affect protein folding in the cell?
Remarkably, the possibility of compact (alpha-helical) structure formation inside the tunnel was experimentally demonstrated 30 years after the original Lim and Spirin’s theoretical suggestion. This was done via several approaches, including FRET (fluorescence resonance energy transfer) [89], probing accessibility (to pegylation) of the engineered cysteines introduced in the growing nascent chain (the length of a stretch of a nascent peptide inside the tunnel has been postulated to be a determinant of helix formation) [90], and by the direct structural studies (see review [91]).
In mid 1970s and 1980s emerging studies also indicated that elongation of translation is a non-uniform process [117-125]. While several reasons for translation non-uniformity were uncovered (including, for example, mRNA structure [118] that may impede the ribosome movement), it became nevertheless clear that the non-uniform synonymous codon usage is one of the key factors modulating translation elongation rates [12-22, 124]. As has been mentioned above, the frequent/optimal codons were as a rule found to be translated more rapidly than the infrequently used ones and vice versa [12-22].
Given this basic knowledge, we then aimed to analyze the codon choice relative to the codon placement at the specific positions in mRNA and examine the relationship between these regions and the encoded protein structures [113-115]. Specifically, we were interested to determine, whether the rare codons can be found at the border regions between identical or different secondary structure elements and/or the domain boundaries. We have postulated that the sequential folding events (such as, e.g., formation of distinct secondary structures and/or tertiary structures, such as domains), which can take place during co-translational folding, might be separated by the rare codons/translational pauses sites [113-115]. Independently, Alistair Brown’s group in the Institute of Genetic at Glasgow University, UK, suggested that the domain folding on the ribosome might be separated by translational pauses and that such regions of slowed translation might serve as interpunctuations during co-translational protein folding [112].
Admittedly, these earlier studies have not been comprehensive enough (partially owing to a lack of sufficient structure and sequence information), nevertheless we have concluded that the locations of rare codons (and their clusters) along mRNAs are highly conserved throughout evolution and they could indeed potentially separate formation of the distinct secondary structure elements, such as, e.g., helices (as has been shown, using single domain proteins, such as cytochromes c, myoglobins and globins) as well as tertiary structure elements, e.g., domains [as has been shown, using multidomain proteins, such as gamma-B and beta-B2 crystallins, phosphoglycerate kinases (PGKs), etc.] [113-115, 126]. Many subsequent and more comprehensive studies [127-136] have identified similar trends at both transition boundaries between different secondary structures (such as, e.g., beta-strand → coil and coil → beta-strand) [134] as well as domains [127, 131-133]. It became also clear that a certain hierarchy exists in the location of rare codon rich regions along mRNA [61, 79, 98, 113-115, 127, 128]. The rarest codons (and their clusters) frequently appeared to encode boundaries of the relatively large units (e.g., domains), whereas less rare codons encode boundaries of the smaller units (e.g., motifs, subdomains). This difference was suggested to reflect the need to provide a more substantial translational delay required for independent folding of the larger units in comparison with the smaller ones [61, 79, 98, 113-115]. It has been also suggested that translational acceleration along the pathway might be equally important as fast forming folding units might require correspondingly faster translation rates [61, 79, 98, 113-115]. The co-translational folding was thus proposed to be considered as a wave process, proceeding through consecutive changes of slow and fast folding phases coupled to slow and fast phases in translation. The so-called codon usage profile (reflecting specific character of codon distribution along mRNA open reading frames) has been proposed to serve as a kinetic guide for co-translational protein folding in the cell [61, 79, 98, 113-115].
Given this knowledge, we then asked, could a link between the specific patterns of synonymous codon usage and protein structures be visualized by comparing the codon usage profiles of the structurally homologous proteins? We hypothesized that if such link exists, then the structurally homologous proteins from different organisms (regardless of the differences in codon usage biases between these organisms) should have similar codon usage profiles [115]. We found that it is indeed the case [115]. Single and multidomain structurally homologous proteins, such as, e.g., cytochromes c, myoglobins, gamma-crystallins, phosphoglycerate kinases (Fig. 2) appeared to have very similar codon usage profiles, revealing conservation of positions of the most rare codons clusters encoding, e.g., domain boundaries [79, 114, 115].
Fig. 2. Conservation of the protein structure is reflected in the conservation of codon usage profile. Backbone/cartoon structures of the phosphoglycerate kinases (PGK) from Escherichia coli (PDB ID 1ZMR) (top), from Saccharomyces cerevisiae yeast (PDB ID 3PGK) (center), and from mouse, Mus musculus (PDB ID 2P9T) (bottom). The respective codon usage profiles are on the right (N- and C-terminal domain boundaries are indicated). Red arrows indicate the conserved cluster of rare codons at the domain boundaries region.
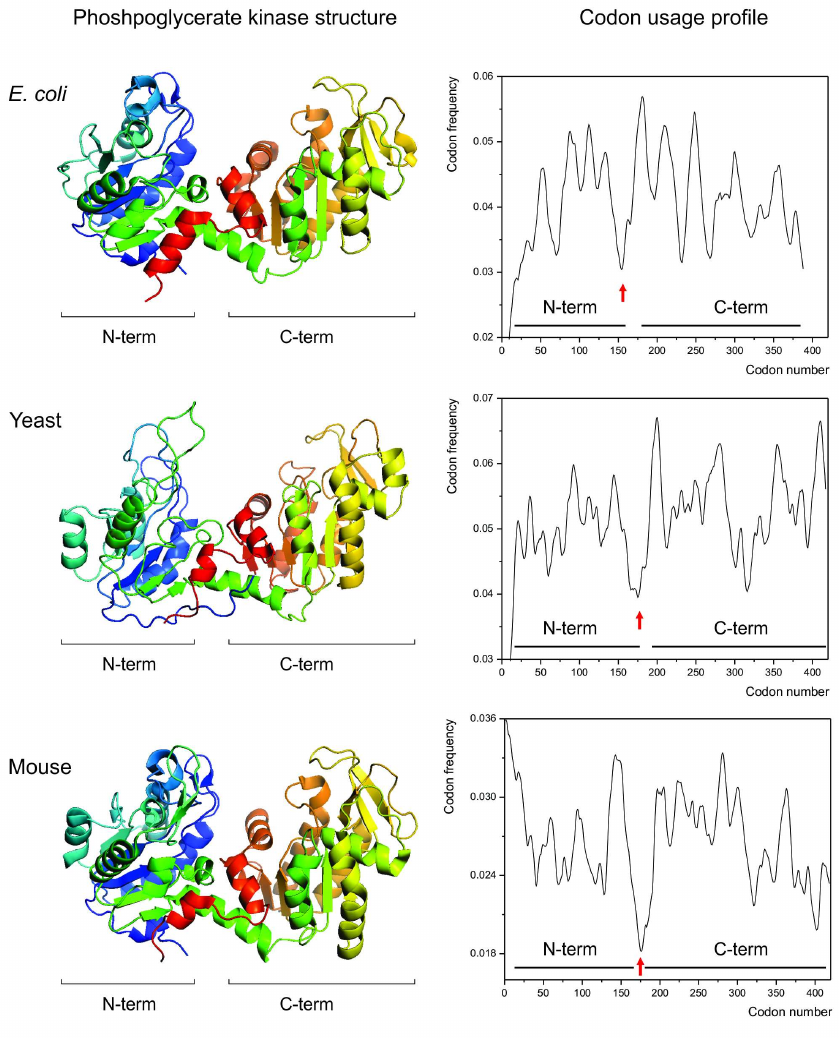
Several subsequent in silico studies, employing for example, chloramphenicol acetyltransferases [131] and ocular lacritins [136] from different organisms provided similar observations.
Importantly, very recent studies also show that the ribosome occupancy profiles are conserved between the structurally and evolutionarily related proteins and protein domains, further supporting the view that kinetics of mRNA translation is evolutionarily conserved between the structurally related proteins to facilitate correct co-translational folding [137].
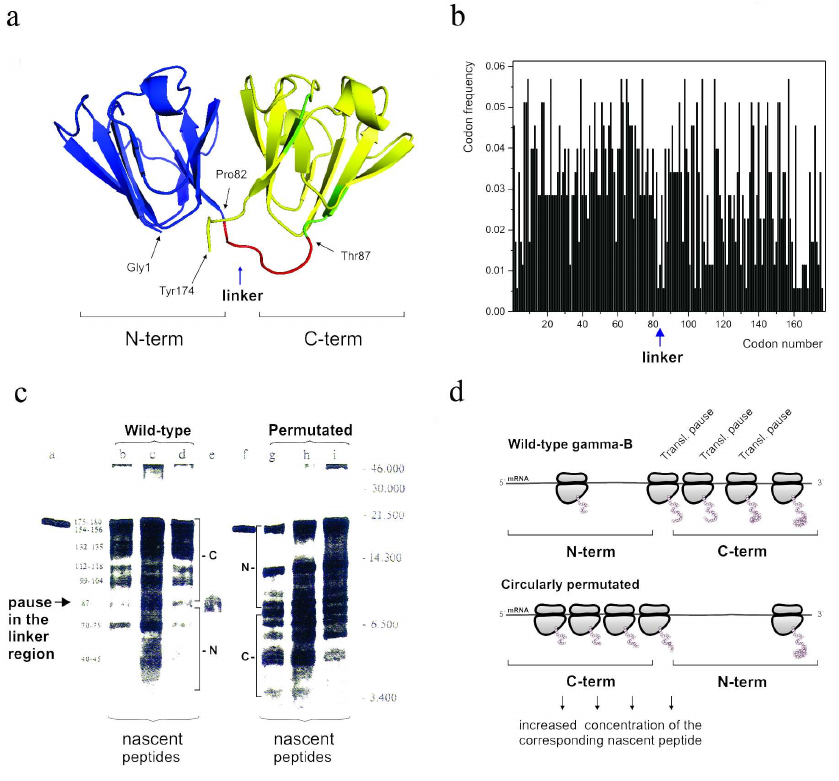
It must be noted, however, that while it is generally accepted now that the frequent codons would accelerate translation and the rare codons would cause a translation pause, earlier attempts aimed at demonstrating that a particular rare codon (or a cluster of rare codons) would determine appearance of the corresponding ribosome-mediated translational pause at a particular place in mRNA appeared to be extremely challenging. Nevertheless, in the early-mid 1990s we were able to show (using both single domain and multidomain proteins) that translation pause sites can separate formation of distinct structural elements, like, e.g., alpha-helices in globins [126] and/or domains in gamma-B crystallin [138]. Interestingly, the in vitro urea-induced equilibrium unfolding and kinetic refolding studies of bovine gamma-B crystallin performed at the same time, revealed that folding of gamma-B crystallin not only proceeds through the sequential structuring of its domains, but is also characterized by the differential kinetics of its domain folding [139]. It was found that the N-terminal domain folds significantly faster than the C-terminal one [139]. Our analysis of bovine gamma-B crystallin synthesis not only confirmed that translation of this protein is a non-uniform process characterized by specific pauses, but has also revealed (by comparing kinetics of translation of the natural bovine gamma-B crystallin and it’s circularly permutated variant with the order of the N- and C-terminal domains exchanged) that the natural N-terminal domain (as a result of non-uniform distribution of fast and rare codons along the mRNA) is translated faster (as was evident by less efficient ribosome pausing in this region) than the C-terminal one [135] (Fig. 3). We have therefore concluded that translation rates can be optimized to tune the synthesis and folding of the nascent polypeptide chains on the ribosome and that the faster folding units could indeed require faster translation times [139].
Fig. 3. The modular (two domain) structure of the gamma-B crystallin and differential kinetics of its domain folding are reflected in the differential codon usage in its domains and non-uniform translation of the protein. a) Backbone/cartoon structure of bovine, Bos taurus, gamma-B crystallin (PDB ID 4GCR); N-terminal (fast folding) domain is in blue and C-terminal (slow folding) domain is in yellow; the linker connecting two domains is in red. b) Codon usage/frequency histogram. The N-terminal domain is (on average) encoded by more frequent codons in comparison with the C-terminal one. There is a clear boundary between the two domains. c) [35S]-Autoradiogram of gamma-B crystallin in vitro translation products (from [138]), a = gamma-B crystallin after 15 min translation; b-d = nascent peptides prepared from polyribosome fractions after 5, 10, and 15 min of gamma-B translation, respectively; e = gamma-B N-terminal domain translated separately; f = gamma circularly permuated protein (gamma-CP) after 15 min translation; g-I = nascent peptides prepared from polyribosome fractions after 5, 10, and 15 min of gamma-CP translation. Major peptides are denoted with numbers according to their lengths (estimated from peptide molecular weights). Arrow indicates the pause in the interdomain (linker region). d) Schematic representation of the (expected) distribution of paused ribosomes in the case of wild-type and circularly permutated gamma-B crystallins. Increased residence time of a ribosome at particular positions along mRNA leads to increased accumulation of the nascent chains of the respective sizes.
The next logical question was: to what extent changes in the synonymous codon usage could affect protein folding in the cell?
In 1999, we have provided one the first observations showing that the synonymous codon substitutions affect ribosome traffic and protein folding during synthesis of a model protein, chloramphenicol acetyltransferase (CAT) [140]. We showed that substitution of sixteen consecutive rare codons in the CAT gene by the frequent ones has led to acceleration of the ribosome traffic through the mutated region in an in vitro translation system and affected specific activity of the enzyme (in comparison with the wild-type protein) [140]. Since specific activity of a given protein could be considered as a measure of its proper folding, we concluded that CAT folding was affected [140].
Many subsequent experiments highlighted significance of the synonymous codon usage for protein folding and showed that synonymous codon substitutions can affect sensitivity of a protein to limited proteolysis [141-143], protein spectroscopic properties [144], aggregation propensity [144-146], specific activity [141, 147], cellular fitness [148], and ultimately can cause diseases [149-152].
In addition, synonymous codon choice has been also suggested to affect efficiency of interaction of the nascent polypeptides with the signal recognition particle [153], thus affecting protein secretion.
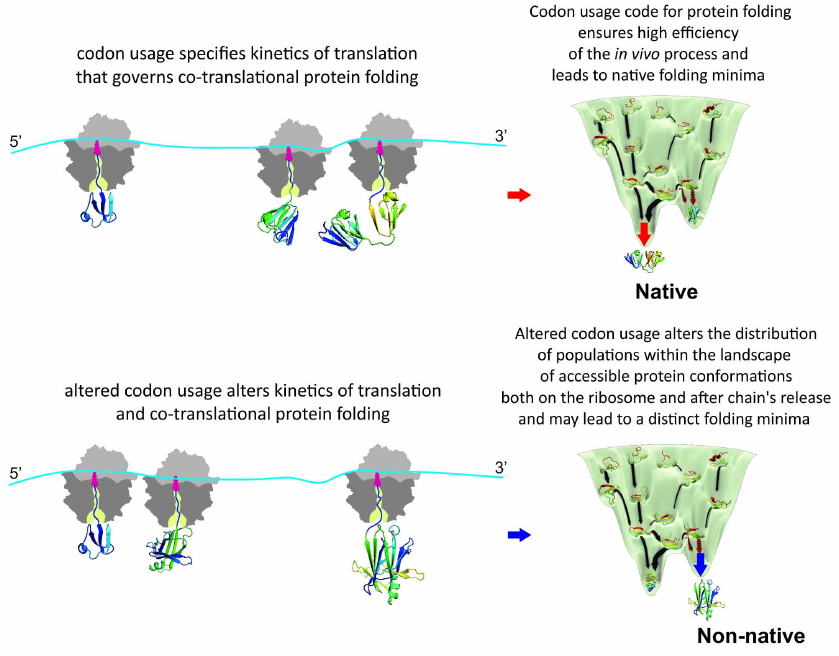
It must be noted the “codon usage code” for protein folding hypothesis [112-115] also postulated that altered kinetics of translation will affect conformation of the ribosome-bound nascent chains in the first place, subsequently potentially also changing final conformation of the released protein and/or altering equilibrium between the different protein conformers, which, in turn, could lead to, e.g., enhanced protein aggregation and/or degradation (co- or post-translational), or change of the protein specific activity and/or function (see reviews [61, 79]). However, direct experimental evidence in support of this notion has been obtained only recently [154]. To investigate how differential usage of synonymous codons affects translation kinetics, co- and post-translational folding, and protein conformation and stability, we further used bovine gamma-B crystallin and analyzed expression of the protein in vivo in Escherichia coli cells and in vitro in a completely reconstituted cell-free translation system. We compared the expression of two variants of gamma-B crystallin, one with the codon usage that would be optimal for protein translation in E. coli (with an mRNA codon usage profile similar to that found in Bos taurus, which was expected to result in more natural translation kinetics) and the other with unaltered codon composition sub-optimal for translation in E. coli [154]. Our analysis of the effects of synonymous codon choice on the translation of gamma-B crystallin mRNAs showed that the codon choice alters local and global translation rates and results in formation of alternative conformations of the protein both in the attached nascent chains and upon release of the protein from the ribosome [154]. Importantly, we showed using real time measurements, which employed fluorescence and FRET, that kinetics of the synthesis and co-translational folding of gamma-B crystallin is indeed altered by the synonymous codon substitutions. Moreover, by using direct structure elucidation approach (2D NMR) we were able to demonstrate, for the first time, considerable structural heterogeneity of the purified, mature synonymous gamma-B crystallin protein variants (which also altered intracellular aggregation propensity of the protein) [154]. We concluded that the synonymous changes introduced in the mRNA altered the distribution of populations within the landscape of accessible protein conformations both on the ribosome and after the chain release [154] (Fig. 4).
These experiments were done in collaboration with Harald Schwalbe from the Johann Wolfgang Goethe-Universität in Frankfurt, Germany and Marina V. Rodnina from the Max Planck Institute for Biophysical Chemistry in Goettingen, Germany, and provided one of the strongest supports to the hypothesis stating that the specific nature of synonymous codon usage along mRNA may indeed serve as a secondary code for protein folding in the cell [154].
Fig. 4. Genetic code governs protein folding in the cell. Appearance of the distinct folding intermediates is linked to the overall and local rate of translation specified by synonymous codon usage. Altered codon usage results in modified translation kinetics and folding events on the ribosome and after polypeptide chain release and may lead to altered final protein conformation and distinct folding minima.
CODON USAGE CODE FOR PROTEIN FOLDING IN THE CELL:
IMPORTANCE
The discovery that the changes of synonymous codons can not only affect the protein expression levels, but also affect protein folding, brought about increased awareness of the scientific community to the impact of synonymous codon usage on the protein function. It became clear that synonymous codon changes could also cause the development of human disease [149-154] and potentially affect safety and efficacy of the recombinant/therapeutic proteins [155-157]. Surprisingly, it was also found that even a single synonymous mutation can be deleterious to protein folding and function [141, 152, 158].
In 2007, Chava Kimchi-Sarfaty, Michael Gottesman and their colleagues demonstrated that a single synonymous mutation in the multidrug resistance 1 (MDR1) gene, encoding P-glycoprotein, could alter protein activity and affect sensitivity of a patient to a plethora of drugs [141]. P-glycoprotein is a transmembrane protein that pumps various drugs out of the cells (see [141] and ref. therein). Under normal conditions it confers resistance to exogenous drug substances and/or body metabolites [141, 149] However, in cancer patients the function of P-glycoprotein (P-gp) can reduce efficacy of chemotherapy treatments and thus P-gp represents an important target for drug delivery purposes [149]. Chava Kimchi-Sarfaty and colleagues showed that substrate specificity of P-glycoprotein is altered by the single synonymous mutation (c.3435>T, ATC>ATT, p.Ile1145Ile) in the gene [141]. It was previously found that plasma and serum blood concentrations of, e.g., cardiac glycoside digoxine, antihistamine drug fexofenadine, μ-opioid receptor agonist loperamide and some other drugs are altered in subjects carrying c.3435>T polymorphism (in comparison with normal individuals) after single or multiple (oral) administration of these drugs, but the exact reason for this difference was not known [149]. The study by Kimchi-Sarfaty and colleagues explained the altered pharmacokinetics of P-gp in the individuals carrying this polymorphism and demonstrated for the first time that the naturally occurring variations in the synonymous codons in a defined gene can give rise to a protein product with the same amino acid sequence but different structural or functional features [141].
Subsequently, in collaboration with Chava Kimchi-Sarfaty, we have demonstrated that a single synonymous mutation, c.459G>A (GTG>GTA, p.Val153Val or Val107Val (Val107 is the amino acid number after the prepro-petide cleavage on secretion)) in the F9 gene (encoding blood coagulation factor FIX) that has been associated with decreased blood coagulation and mild haemophilia B in the affected individuals, alters FIX synthesis and affects its conformation resulting in the decreased extracellular protein levels [152]. As such, we were able to determine pathogenic basis for the single synonymous mutation in the F9 gene associated with haemophilia B [152]. This case was puzzling for researchers, who discovered it in 2008 [159], as it could not be explained by the altered mRNA properties (mRNA levels, splicing/exon skipping or retention of introns, or stability), a common expected cause [150, 151, 160-163] of many diseases associated with synonymous mutations, before it became clear that synonymous mutations could affect protein biogenesis.
In addition, and also in collaboration with Chava Kimchi-Sarfaty, we have recently demonstrated that a single synonymous mutation, c.354G>A (CCG>CCA, p.Pro118Pro) in ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13), a large multi-domain secreted protein that regulates thrombogenesis by cleavage of an adhesive blood glycoprotein, von Willebrand factor (VWF) can confer enhanced specific activity to the enzyme [158]. This substitution is a naturally occurring variant in the human population (allele frequency: 0.026 (1000 Genomes), 0.0627 [The Exome Aggregation Consortium (ExAC)] [158], and thus far has not been associated with a disease phenotype [158, 164]. However, c.354G>A ADAMTS13 variant to date represents a truly unique example, as it produces a substantial positive contribution to the protein specific activity [158]. Whether this mutation offers any evolutionary advantage to the affected individuals remains to be established.
The finding that synonymous codon changes can represent a risk factor for proper protein function (and affect protein structure) also adds a novel layer of complexity to the process of manufacturing of recombinant proteins, including protein therapeutics. Currently, biopharmaceutical industry takes advantage of an array of available computational tools, which take into account synonymous codon usage bias, to redesign genes for enhanced expression (see review [156]). However, these tools typically focus on the translation efficiency (and usually involve massive substitutions of the majority of infrequently used codons with synonymous frequently used ones) and have limited (if any) capacity to evaluate the effect of synonymous codon usage on protein folding [156]. However, as mentioned above, maximizing the speed and output of translation may put conflicting demands on the protein synthesis machinery, resulting in improper protein folding. Moreover, a growing number of reports show that even minor changes in the protein structure can result in altered immunogenicity of the protein therapeutics and lead to the development of neutralizing antibodies [155, 165]. These findings represent an additional risk factor during manufacturing of protein therapeutics. At present, assessment of the potential for immunogenicity in protein therapeutics represents another unmet need in biotechnology industry and in clinical practice [165].
Obviously, the development of better codon optimization strategies is required. Several companies, including, for example DAPCEL, Inc. [156], are now utilizing the available knowledge for the development of better synonymous gene optimization strategies aimed at production (in any desired host organism) of correctly folded, soluble proteins.
However, much work has yet to be done. There is yet an incomplete understanding of the exact impact of both single and multiple synonymous mutations on protein folding and function. Nevertheless, despite the lack of understanding of many details, it became obvious that synonymous codon usage provides a secondary code for the protein folding in vivo and fine-tunes the co-translational protein folding by allowing proteins to circumvent deep kinetic traps during synthesis and thus ensuring rapid and efficient acquisition of the protein structure in the cell. It is thus becoming evident that natural selection simultaneously exploits numerous possibilities to optimize expression of the genetic information.
CONCLUDING REMARKS
This review is dedicated to the memory of Alexander Sergeevich Spirin and on occasion of his 90th birthday anniversary and in part reflects his contribution to the development of the field of co-translational protein folding.
My interest in protein synthesis, as I believe the interest of many students and researchers in the Department of Molecular Biology at Moscow State University, which Prof. Spirin headed from 1972 to 2012, was largely triggered by his lectures on the ribosome structure and the mechanism of protein synthesis. I had a chance to attend these lectures back in 1984, as an undergraduate student in the Department. These lectures were truly inspiring, stimulating, and prompting numerous questions. I was fortunate to continue my graduate and post-graduate work in the same Department in the groups of Prof. Vladimir V. Yurkevich and subsequently Prof. Igor A. Krasheninnikov, also working in close collaboration with Prof. Spirin. I have had also a chance to spend substantial time in the Prof. Spirin’s laboratory at the Institute of Protein Research, in Pushchino (in early-mid 1990s) conducting work on co-translational folding of globin. This experience was invaluable for shaping my scientific views and attitude for research.
Acknowledgments. This work would not be possible without the original contribution of Ivan Adzhubei and Igor A. Krasheninnikov, further support from Slava Kolb, Aigar Kommer, Valentin M. Stepanov, Lev P. Ovchinnikov, and Alexander S. Spirin, followed by collaborations with Rainer Jaenicke, Claude Reiss and more recently with Chava Kimchi-Sarfaty, Harald Schwalbe and Marina V. Rodnina.
I am indebted to all my teachers, colleagues, and collaborators for their extremely generous and inspiring discussions and invaluable contributions. I also apologize to those whose work or original publications could not be cited in this short review article.
Funding. In recent years, the work in my laboratory was financially supported by the Human Frontier Science Program Organization [HFSP grant #RGP0024/2010], the American Heart Association [AHA grant 13GRNT17070025], the National Institutes of Health [NIH grants HL121779; HL151392 and GM128981], the Center for Gene Regulation in Health and Disease (GRHD) at CSU, and the biotechnology company, DAPCEL, Inc.
Ethics declarations. I am co-founder and Chief Scientific Officer of DAPCEL, Inc., a biotechnology company that develops innovative approaches for gene redesign for protein production in any desired host organism. This article does not contain any studies with human participants or animals performed by the author.
REFERENCES
1.Crick, F. H., Barnett, L., Brenner, S., and
Watts-Tobin, R. J. (1961) General nature of the genetic code for
proteins, Nature, 192, 1227-1232.
2.Nirenberg, M., and Leder, P. (1964) RNA codewords
and protein synthesis. The effect of trinucleotides upon the binding of
sRNA to ribosomes, Science, 145, 1399-1407.
3.Leder, P., and Nirenberg, M. (1964) RNA codewords
and protein synthesis. II. nucleotide sequence of a valine RNA
codeword, Proc. Natl. Acad. Sci. USA, 52, 420-427.
4.Bernfield, M. R., and Nirenberg, M. W. (1965) RNA
codewords and protein synthesis. The nucleotide sequences of multiple
codewords for phenylalanine, serine, leucine, and proline,
Science, 147, 479-484.
5.Trupin, J. S., Rottman, F. M., Brimacombe, R. L.,
Leder, P., Bernfield, M. R., and Nirenberg, M. W. (1965) RNA codewords
and protein synthesis, VI. On the nucleotide sequences of degenerate
codeword sets for isoleucine, tyrosine, asparagine, and lysine,
Proc. Natl. Acad. Sci. USA, 53, 807-811.
6.Nirenberg, M., Leder, P., Bernfield, M.,
Brimacombe, R., Trupin, J., et al. (1965) RNA codewords and protein
synthesis, VII. On the general nature of the RNA code, Proc. Natl.
Acad. Sci. USA, 53, 1161-1168.
7.Brimacombe, R., Trupin, J., Nirenberg, M., Leder,
P., Bernfield, M., and Jaouni, T. (1965) RNA codewords and protein
synthesis, 8. Nucleotide sequences of synonym codons for arginine,
valine, cysteine, and alanine, Proc. Natl. Acad. Sci. USA,
54, 954-960.
8.Söll, D., Ohtsuka, E., Jones, D. S., Lohrmann,
R., Hayatsu, H., et al. (1965) Studies on polynucleotides, XLIX.
Stimulation of the binding of aminoacyl-sRNA’s to ribosomes by
ribotrinucleotides and a survey of codon assignments for 20 amino
acids, Proc. Natl. Acad. Sci. USA, 54, 1378-1385.
9.Goel, N. S., Rao, G. S., Ycas, M., Bremermann, H.
J., and King, L. (1972) A method for calculating codon frequencies in
DNA, J. Theor. Biol., 35, 399-457.
10.Grantham, R., Gautier, C., Gouy, M., Mercier, R.,
and Pavé, A. (1980) Codon catalog usage and the genome
hypothesis, Nucleic Acids Res., 8, r49-r62.
11.Grantham, R., Gautier, C., and Gouy, M. (1980)
Codon frequencies in 119 individual genes confirm consistent choices of
degenerate bases according to genome type, Nucleic Acids Res.,
8, 1893-1912.
12.Ikemura, T. (1981) Correlation between the
abundance of Escherichia coli transfer RNAs and the occurrence of the
respective codons in its protein genes, J. Mol. Biol.,
146, 1-21.
13.Ikemura, T. (1982) Correlation between the
abundance of yeast transfer RNAs and the occurrence of the respective
codons in protein genes. Differences in synonymous codon choice
patterns of yeast and Escherichia coli with reference to the abundance
of isoaccepting transfer RNAs, J. Mol. Biol., 158,
573-597.
14.Bennetzen, J. L., and Hall, B. D. (1982) Codon
selection in yeast, J. Biol. Chem., 257, 3026-3031.
15.Hastings, K. E. M., and Emerson, C. P. Jr. (1983)
Codon usage in muscle genes and liver genes, J. Mol. Evol.,
19, 214-218.
16.Ikemura, T. (1985) Codon usage and tRNA content
in unicellular and multicellular organisms, Mol. Biol. Evol.,
2, 13-34.
17.Sharp, P. M., Tuohy, T. M., and Mosurski, K. R.
(1986) Codon usage in yeast: cluster analysis clearly differentiates
highly and lowly expressed genes, Nucleic Acids Res., 14,
5125-5143.
18.Sharp, P. M., and Li, W. H. (1987) The Codon
Adaptation Index – a measure of directional synonymous codon
usage bias, and its potential applications, Nucleic Acids Res.,
15, 1281-1295.
19.Sharp, P. M., Cowe, E., Higgins, D. G., Shields,
D. C., Wolfe, K. H., and Wright, F. (1988) Codon usage patterns in
Escherichia coli, Bacillus subtilis, Saccharomyces
cerevisiae, Schizosaccharomyces pombe, Drosophila
melanogaster and Homo sapiens; a review of the considerable
within-species diversity, Nucleic Acids Res., 16,
8207-8211.
20.Sørensen, M.A., Kurland, C. G., and
Pedersen, S. (1989) Codon usage determines translation rate in
Escherichia coli, J. Mol. Biol., 207, 365-377.
21.Andersson, S. G., and Kurland, C. G. (1990) Codon
preferences in free-living microorganisms, Microbiol. Rev.,
54, 198-210.
22.Kurland, C. G. (1991) Codon bias and gene
expression, FEBS Lett., 285, 165-169.
23.Kramer, E. B., and Farabaugh, P. J. (2007) The
frequency of translational misreading errors in E. coli is
largely determined by tRNA competition, RNA, 13,
87-96.
24.Zaher, H. S., and Green, R. (2009) Fidelity at
the molecular level: lessons from protein synthesis, Cell,
136, 746-762.
25.Kramer, E. B., Vallabhaneni, H., Mayer, L. M.,
and Farabaugh, P. J. (2010) A comprehensive analysis of translational
missense errors in the yeast Saccharomyces cerevisiae,
RNA, 16, 1797-1808.
26.Ribas de Pouplana, L., Santos, M. A., Zhu, J. H.,
Farabaugh, P. J., and Javid, B. (2014) Protein mistranslation: friend
or foe? Trends. Biochem. Sci., 39, 355-362.
27.Huang, Y., Koonin, E. V., Lipman, D. J., and
Przytycka, T. M. (2009) Selection for minimization of translational
frameshifting errors as a factor in the evolution of codon usage,
Nucleic Acids Res., 37, 6799-6810.
28.Savir, Y., and Tlusty, T. (2013) The ribosome as
an optimal decoder: a lesson in molecular recognition, Cell,
153, 471-479.
29.Dana, A., and Tuller, T. (2014) The effect of
tRNA levels on decoding times of mRNA codons, Nucleic Acids
Res., 42, 9171-9181.
30.Bonekamp, F., Andersen, H. D., Christensen, T.,
and Jensen, K. F. (1985) Codon-defined ribosomal pausing in
Escherichia coli detected by using the pyrE attenuator to probe
the coupling between transcription and translation, Nucleic Acids
Res., 13, 4113-4123.
31.Curran, J. F., and Yarus, M. (1989) Rates of
aminoacyl-tRNA selection at 29 sense codons in vivo, J. Mol.
Biol., 209, 65-77.
32.Bulmer, M. (1987) Coevolution of codon usage and
transfer RNA abundance, Nature, 325, 728-730.
33.Duret, L. (2000) tRNA gene number and codon usage
in the C. elegans genome are co-adapted for optimal translation
of highly expressed genes, Trends Genet., 16,
287-289.
34.Li, W. H. (1987) Models of nearly neutral
mutation with particular implications for nonrandom usage of synonymous
codons, J. Mol. Evol., 24, 337-345.
35.Shields, D. C. (1990) Switches in
species-specific codon preferences: the influence of mutation biases,
J. Mol. Evol., 31, 71-80.
36.Bulmer, M. (1991) The selection-mutation-drift
theory of synonymous codon usage, Genetics, 129,
897-907.
37.Presnyak, V., Alhusaini, N., Chen, Y. H., Martin,
S., Morris, N., et al. (2015) Codon optimality is a major determinant
of mRNA stability, Cell, 160, 1111-1124, doi:
10.1016/j.cell.2015.02.029.
38.Boël, G., Letso, R., Neely, H., Price, W.
N., Wong, K. H., et al. (2016) Codon influence on protein expression in
E. coli correlates with mRNA levels, Nature, 529,
358-363.
39.Mishima, Y., and Tomari, Y. (2016) Codon usage
and 3′-UTR length determine maternal mRNA stability in zebrafish,
Mol. Cell, 61, 874-885.
40.Bazzini, A. A., Del Viso, F., Moreno-Mateos, M.
A., Johnstone, T. G., Vejnar, C. E., et al. (2016) Codon identity
regulates mRNA stability and translation efficiency during the
maternal-to-zygotic transition, EMBO J., 35,
2087-2103.
41.Radhakrishnan, A., Chen, Y. H., Martin, S.,
Alhusaini, N., Green, R., and Coller, J. (2016) The DEAD-box protein
Dhh1p couples mRNA decay and translation by monitoring codon
optimality, Cell, 167, 122-132.
42.Hia, F., Yang, S. F., Shichino, Y., Yoshinaga,
M., Murakawa, Y., et al. (2019) Codon bias confers stability to human
mRNAs, EMBO Rep., 20, e48220.
43.Wu, Q., Medina, S. G., Kushawah, G., DeVore, M.
L., Castellano, L. A., et al. (2019) Translation affects mRNA stability
in a codon-dependent manner in human cells, Elife, 8,
e45396.
44.Medina-Muñoz, S. G., Kushawah, G.,
Castellano, L. A., Diez, M., DeVore, M. L., et al. (2021) Crosstalk
between codon optimality and cis-regulatory elements dictates mRNA
stability, Genome Biol., 22, 14.
45.Anfinsen, C. B. (1973) Principles that govern the
folding of protein chains, Science, 181, 223-230.
46.Jaenicke, R. (1991) Protein folding: local
structures, domains, subunits, and assemblies, Biochemistry,
30, 3147-3161.
47.Fersht, A. R. (2008) From the first protein
structures to our current knowledge of protein folding: delights and
skepticisms, Nat. Rev. Mol. Cell Biol., 9, 650-654.
48.Bartlett, A. I., and Radford, S. E. (2009) An
expanding arsenal of experimental methods yields an explosion of
insights into protein folding mechanisms, Nat. Struct. Mol.
Biol., 16, 582-588.
49.Finkelstein, A. V. (2018) 50+ years of protein
folding, Biochemistry (Moscow), 83 (Suppl. 1),
S3-S18.
50.Abaskharon, R. M., and Gai, F. (2016) Meandering
down the energy landscape of protein folding: are we there yet?
Biophys. J., 110, 1924-1932.
51.Ferina, J., and Daggett, V. (2019) Visualizing
protein folding and unfolding, J. Mol. Biol., 431,
1540-1564.
52.Cowie, D. B., Spiegelman, S., Roberts, R. B., and
Duerksen, J. D. (1961) Ribosome-bound β-galactosidase, Proc.
Natl. Acad. Sci. USA, 47, 114-122.
53.Zipser, D., and Perrin, D. (1963) Complementation
on ribosomes, Cold Spring Harb. Symp. Quant. Biol., 28,
533-537.
54.Kiho, Y., and Rich, A. (1964) Induced enzyme
formed on bacterial polyribosomes, Proc. Natl. Acad. Sci. USA,
51, 111-118.
55.Hamlin, J., and Zabin, I. (1972)
β-Galactosidase: immunological activity of ribosome-bound, growing
polypeptide chains, Proc. Natl. Acad. Sci. USA, 69,
412-416.
56.Bergman, L. W., and Kuehl, W. M. (1979) Formation
of intermolecular disulfide bonds on nascent immunoglobulin
polypeptides, J. Biol. Chem., 254, 5690-5694.
57.Bergman, L. W., and Kuehl, W. M. (1979) Formation
of an intrachain disulfide bond on nascent immunoglobulin light chains,
J. Biol. Chem., 254, 8869-8876.
58.Bergman, L. W., and Kuehl, W. M. (1979)
Co-translational modification of nascent immunoglobulin heavy and light
chains, J. Supramol. Struct., 11, 9-24.
59.Lim, V. I., and Spirin, A. S. (1985)
Stereochemistry of the transpeptidation reaction in the ribosome. The
ribosome generates an alpha-helix in the synthesis of the protein
polypeptide chain, Dokl. Akad. Nauk SSSR, 280,
235-239.
60.Lim, V. I., and Spirin, A. S. (1986)
Stereochemical analysis of ribosomal transpeptidation. Conformation of
nascent peptide, J. Mol. Biol., 188, 565-574.
61.Komar, A. A. (2019) Synonymous codon usage
– a guide for co-translational protein folding in the cell,
Mol. Biol. (Mosk), 53, 883-898.
62.Noé, F., De Fabritiis, G., and Clementi,
C. (2020) Machine learning for protein folding and dynamics, Curr.
Opin. Struct. Biol., 60, 77-84.
63.Gao, W., Mahajan, S. P., Sulam, J., and Gray, J.
J. (2020) Deep learning in protein structural modeling and design,
Patterns (NY), 1, 100142.
64.Senior, A. W., Evans, R., Jumper, J.,
Kirkpatrick, J., Sifre, L., et al. (2020) Improved protein structure
prediction using potentials from deep learning, Nature,
577, 706-710.
65.Lindberg, M. O., and Oliveberg, M. (2007)
Malleability of protein folding pathways: a simple reason for complex
behaviour, Curr. Opin. Struct. Biol., 17, 21-29.
66.Englander, S. W., and Mayne, L. (2014) The nature
of protein folding pathways, Proc. Natl. Acad. Sci. USA,
111, 15873-15880.
67.Englander, S. W., Mayne, L., Kan, Z. Y., and Hu,
W. (2016) Protein folding-how and why: by hydrogen exchange, fragment
separation, and mass spectrometry, Annu. Rev. Biophys.,
45, 135-152.
68.Levinthal, C. (1969). How to fold graciously, in
Mossbauer Spectroscopy in Biological Systems, Proceedings of a
Meeting held at Allerton House, Monticello, Illinois (Debrunner, P.,
Tsibris, J. C. M., and Münck, E., eds.) University of Illinois
Press, Urbana, p. 22.
69.Gopan, G., Gruebele, M., and Rickard, M. (2020)
In-cell protein landscapes: making the match between theory, simulation
and experiment, Curr. Opin. Struct. Biol., 66,
163-169.
70.Gershenson, A., Gosavi, S., Faccioli, P., and
Wintrode, P. L. (2020) Successes and challenges in simulating the
folding of large proteins, J. Biol. Chem., 295,
15-33.
71.Chow, M. K., Amin, A. A., Fulton, K. F.,
Fernando, T., Kamau, L., et al. (2006) The REFOLD database: a tool for
the optimization of protein expression and refolding, Nucleic Acids
Res., 34 (Database issue), D207-212.
72.Mizutani, H., Sugawara, H., Buckle, A. M.,
Sangawa, T., Miyazono, K. I., et al. (2017) REFOLDdb: a new and
sustainable gateway to experimental protocols for protein refolding,
BMC Struct. Biol., 17, 4.
73.Hartl, F. U., and Hayer-Hartl, M. (2009)
Converging concepts of protein folding in vitro and in
vivo, Nat. Struct. Mol. Biol., 16, 574-581.
74.Hingorani, K. S., and Gierasch, L. M. (2014)
Comparing protein folding in vitro and in vivo:
foldability meets the fitness challenge, Curr. Opin. Struct.
Biol., 24, 81-90.
75.Balchin, D., Hayer-Hartl, M., and Hartl, F. U.
(2016) In vivo aspects of protein folding and quality control,
Science, 353, aac4354.
76.Gruebele, M., Dave, K., and Sukenik, S. (2016)
Globular protein folding in vitro and in vivo, Annu.
Rev. Biophys., 45, 233-251.
77.Dahiya, V., and Buchner, J. (2019) Functional
principles and regulation of molecular chaperones, Adv. Protein.
Chem. Struct. Biol., 114, 1-60.
78.Jayaraj, G. G., Hipp, M. S., and Hartl, F. U.
(2019) Functional modules of the proteostasis network, Cold Spring
Harb. Perspect. Biol., 12, a033951.
79.Komar, A. A. (2009) A pause for thought along the
co-translational folding pathway, Trends Biochem. Sci.,
34, 16-24.
80.Gloge, F., Becker, A. H., Kramer, G., and Bukau,
B. (2014) Co-translational mechanisms of protein maturation, Curr.
Opin. Struct. Biol., 24, 24-33.
81.Thommen, M., Holtkamp, W., and Rodnina, M. V.
(2017) Co-translational protein folding: progress and methods, Curr.
Opin. Struct. Biol., 42, 83-89.
82.Komar, A. A. (2018) Unraveling co-translational
protein folding: concepts and methods, Methods, 137,
71-81.
83.Williams, N. K., and Dichtl, B. (2018)
Co-translational control of protein complex formation: a fundamental
pathway of cellular organization? Biochem. Soc. Trans.,
46, 197-206.
84.Waudby, C. A., Dobson, C. M., and Christodoulou,
J. (2019) Nature and regulation of protein folding on the ribosome,
Trends Biochem. Sci., 44, 914-926.
85.Jha, S., and Komar, A. A. (2011) Birth, life and
death of nascent polypeptide chains, Biotechnol. J., 6,
623-640.
86.Das, D., Das, A., Samanta, D., Ghosh, J.,
Dasgupta, S., et al. (2008) Role of the ribosome in protein folding,
Biotechnol. J., 3, 999-1009.
87.Voisset, C., Saupe, S. J., and Blondel, M. (2011)
The various facets of the protein-folding activity of the ribosome,
Biotechnol. J., 6, 668-673.
88.Banerjee, D., and Sanyal, S. (2014) Protein
folding activity of the ribosome (PFAR) – a target for antiprion
compounds, Viruses, 6, 3907-3924.
89.Woolhead, C. A., McCormick, P. J., and Johnson,
A. E. (2004) Nascent membrane and secretory proteins differ in
FRET-detected folding far inside the ribosome and in their exposure to
ribosomal proteins, Cell, 116, 725-736.
90.Lu, J., and Deutsch, C. (2005) Folding zones
inside the ribosomal exit tunnel, Nat. Struct. Mol. Biol.,
12, 1123-1129.
91.Wilson, D. N., and Beckmann, R. (2011) The
ribosomal tunnel as a functional environment for nascent polypeptide
folding and translational stalling, Curr. Opin. Struct. Biol.,
21, 274-282.
92.Tu, L., and Deutsch, C. (2017) Determinants of
helix formation for a Kv1.3 transmembrane segment inside the ribosome
exit tunnel, J. Mol. Biol., 429, 1722-1732.
93.Gilbert, R. J., Fucini, P., Connell, S., Fuller,
S. D., Nierhaus, K. H., et al. (2004) Three-dimensional structures of
translating ribosomes by Cryo-EM, Mol. Cell, 14,
57-66.
94.Kosolapov, A., and Deutsch, C. (2009) Tertiary
interactions within the ribosomal exit tunnel, Nat. Struct. Mol.
Biol., 16, 405-411.
95.Tu, L., Khanna, P., and Deutsch, C. (2014)
Transmembrane segments form tertiary hairpins in the folding vestibule
of the ribosome, J. Mol. Biol., 426, 185-198.
96.Holtkamp, W., Kokic, G., Jäger, M.,
Mittelstaet, J., Komar, A. A., and Rodnina, M. V. (2015)
Cotranslational protein folding on the ribosome monitored in real time,
Science, 350, 1104-1107.
97.Chen, X., Rajasekaran, N., Liu, K., and Kaiser,
C. M. (2020) Synthesis runs counter to directional folding of a nascent
protein domain, Nat. Commun., 11, 5096.
98.Komar, A. A. (2018) The Yin and Yang of codon
usage, Hum. Mol. Genet., 25(R2), R77-R85.
99.Komar, A. A. (2007) SNPs, silent but not
invisible, Science, 315, 466-467.
100.Tsai, C. J., Sauna, Z. E., Kimchi-Sarfaty, C.,
Ambudkar, S. V., Gottesman, M. M., and Nussinov, R. (2008) Synonymous
mutations and ribosome stalling can lead to altered folding pathways
and distinct minima, J. Mol. Biol., 383, 281-291.
101.Zhang, G., and Ignatova, Z. (2011) Folding at
the birth of the nascent chain: coordinating translation with
co-translational folding, Curr. Opin. Struct. Biol., 21,
25-31.
102.O’Brien, E. P., Ciryam, P., Vendruscolo,
M., and Dobson, C. M. (2014) Understanding the influence of codon
translation rates on cotranslational protein folding, Acc. Chem.
Res., 47, 1536-1544.
103.Chaney, J. L., and Clark, P. L. (2015) Roles
for synonymous codon usage in protein biogenesis, Annu. Rev.
Biophys., 44, 143-166.
104.Jacobson, G. N., and Clark, P. L. (2016)
Quality over quantity: optimizing co-translational protein folding with
non-“optimal” synonymous codons, Curr. Opin. Struct.
Biol., 38, 102-110.
105.Sharma, A. K., and O’Brien, E. P. (2018)
Non-equilibrium coupling of protein structure and function to
translation-elongation kinetics, Curr. Opin. Struct. Biol.,
49, 94-103.
106.Samatova, E., Daberger, J., Liutkute, M., and
Rodnina, M. V. (2021) Translational control by ribosome pausing in
bacteria: how a non-uniform pace of translation affects protein
production and folding, Front. Microbiol., 11,
619430.
107.Liu, Y., Yang, Q., and Zhao, F. (2021)
Synonymous but not silent: the codon usage code for gene expression and
protein folding, Annu. Rev. Biochem., 90, 375-401, doi:
10.1146/annurev-biochem-071320-112701.
108.Pelham, H. R., and Jackson, R. J. (1976) An
efficient mRNA-dependent translation system from reticulocyte lysates,
Eur. J. Biochem., 67, 247-256.
109.Kolb, V. A., Makeyev, E. V., and Spirin, A. S.
(1994) Folding of firefly luciferase during translation in a cell-free
system, EMBO J., 13, 3631-3637.
110.Komar, A. A., Kommer, A., Krasheninnikov, I.
A., and Spirin, A. S. (1993) Cotranslational heme binding to nascent
globin chains, FEBS Lett., 326, 261-263.
111.Komar, A. A., Kommer, A., Krasheninnikov, I.
A., and Spirin, A. S. (1997) Cotranslational folding of globin, J.
Biol. Chem., 272, 10646-10651.
112.Purvis, I. J., Bettany, A. J., Santiago, T. C.,
Coggins, J. R., Duncan, K., et al. (1987) The efficiency of folding of
some proteins is increased by controlled rates of translation in
vivo. A hypothesis, J. Mol. Biol., 193, 413-417.
113.Krasheninnikov, I. A., Komar, A. A., and
Adzhubeĭ, I. A. (1988) Role of the rare codon clusters in
defining the boundaries of polypeptide chain regions with identical
secondary structures in the process of co-translational folding of
proteins [in Russian], Dokl. Akad. Nauk SSSR, 303,
995-999.
114.Krasheninnikov, I. A., Komar, A. A., and
Adzhubeĭ, I. A. (1989) Frequency of using codons in mRNA and
coding of the domain structure of proteins [in Russian], Dokl. Akad.
Nauk. SSSR, 305, 1006-1012.
115.Krasheninnikov, I. A., Komar, A. A., and
Adzhubeĭ, I. A. (1989) Role of the code redundancy determining
cotranslational protein folding, Biokhimiia, 5,
187-200.
116.Heinemann, U., and Hahn, M. (1995) Circular
permutation of polypeptide chains: implications for protein folding and
stability, Prog. Biophys. Mol. Biol., 64, 121-143.
117.Protzel, A., and Morris, A. J. (1974) Gel
chromatographic analysis of nascent globin chains. Evidence of
nonuniform size distribution, J. Biol. Chem., 249,
4594-4600.
118.Chaney, W. G., and Morris, A. J. (1978)
Nonuniform size distribution of nascent peptides: the role of messenger
RNA, Arch. Biochem. Biophys., 191, 734-741.
119.Lizardi, P. M., Mahdavi, V., Shields, D., and
Candelas, G. (1979) Discontinuous translation of silk fibroin in a
reticulocyte cell-free system and in intact silk gland cells, Proc.
Natl. Acad. Sci. USA, 76, 6211-6215.
120.Randall, L. L., Josefsson, L. G., and Hardy, S.
J. (1980) Novel intermediates in the synthesis of maltose-binding
protein in Escherichia coli, Eur. J. Biochem.,
107, 375-379.
121.Abraham, A. K., and Pihl, A. (1980) Variable
rate of polypeptide chain elongation in vitro. Effect of
spermidine, Eur. J. Biochem., 106, 257-262.
122.Varenne, S., Knibiehler, M., Cavard, D.,
Morlon, J., and Lazdunski, C. (1982) Variable rate of polypeptide chain
elongation for colicins A, E2 and E3, J. Mol. Biol., 159,
57-70.
123.Candelas, G., Candelas, T., Ortiz, A., and
Rodríguez, O. (1983) Translational pauses during a spider
fibroin synthesis, Biochem. Biophys. Res. Commun., 116,
1033-1038.
124.Varenne, S., Buc, J., Lloubes, R., and
Lazdunski, C. (1984) Translation is a non-uniform process. Effect of
tRNA availability on the rate of elongation of nascent polypeptide
chains, J. Mol. Biol., 180, 549-576.
125.Wolin, S. L., and Walter, P. (1988) Ribosome
pausing and stacking during translation of a eukaryotic mRNA, EMBO
J., 7, 3559-3569.
126.Krasheninnikov, I. A., Komar, A. A., and
Adzhubeĭ, I. A. (1991) Nonuniform size distribution of nascent
globin peptides, evidence for pause localization sites, and a
cotranslational protein-folding model, J. Protein. Chem.,
10, 445-454.
127.Thanaraj, T. A., and Argos, P. (1996)
Ribosome-mediated translational pause and protein domain organization,
Protein Sci., 5, 1594-1612.
128.Thanaraj, T. A., and Argos, P. (1996) Protein
secondary structural types are differentially coded on messenger RNA,
Protein Sci., 5, 1973-1983.
129.Adzhubei, A. A., Adzhubei, I. A.,
Krasheninnikov, I. A., and Neidle, S. (1996) Non-random usage of
“degenerate” codons is related to protein three-dimensional
structure, FEBS Lett., 399, 78-82.
130.Oresic, M., and Shalloway, D. (1998) Specific
correlations between relative synonymous codon usage and protein
secondary structure, J. Mol. Biol., 281, 31-48.
131.Widmann, M., Clairo, M., Dippon, J., and
Pleiss, J. (2008) Analysis of the distribution of functionally relevant
rare codons, BMC Genomics, 9, 207.
132.Clarke, T. F. 4th, and Clark, P. L. (2008) Rare
codons cluster, PLoS One, 3, e3412.
133.Chartier, M., Gaudreault, F., and Najmanovich,
R. (2012) Large-scale analysis of conserved rare codon clusters
suggests an involvement in co-translational molecular recognition
events, Bioinformatics, 28, 1438-1445.
134.Ma, X. X., Wang, Y. N., Cao, X. A., Li, X. R.,
Liu, Y. S., et al. (2018) The effects of codon usage on the formation
of secondary structures of nucleocapsid protein of peste des petits
ruminants virus, Genes Genomics, 40, 905-912.
135.Newaz, K., Wright, G., Piland, J., Li, J.,
Clark, P. L., et al. (2020) Network analysis of synonymous codon usage,
Bioinformatics, 36, 4876-4884.
136.McKown, R. L., Raab, R. W., Kachelries, P.,
Caldwell, S., and Laurie, G. W. (2013) Conserved regional 3′
grouping of rare codons in the coding sequence of ocular prosecretory
mitogen lacritin, Invest. Ophthalmol. Vis. Sci., 54,
1979-1987.
137.Nissley, D. A., Carbery, A., Chonofsky, M., and
Deane, C. M. (2021) Ribosome occupancy profiles are conserved between
structurally and evolutionarily related yeast domains,
Bioinformatics, doi: 10.1093/bioinformatics/btab020.
138.Komar, A. A., and Jaenicke, R. (1995) Kinetics
of translation of gamma B crystallin and its circularly permutated
variant in an in vitro cell-free system: possible relations to
codon distribution and protein folding, FEBS Lett., 376,
195-198.
139.Rudolph, R., Siebendritt, R., Nesslaŭer,
G., Sharma, A. K., and Jaenicke, R. (1990) Folding of an all-beta
protein: independent domain folding in gamma II-crystallin from calf
eye lens, Proc. Natl. Acad. Sci. USA, 87, 4625-4629.
140.Komar, A. A., Lesnik, T., and Reiss, C. (1999)
Synonymous codon substitutions affect ribosome traffic and protein
folding during in vitro translation, FEBS Lett.,
462, 387-391.
141.Kimchi-Sarfaty, C., Oh, J. M., Kim, I. W.,
Sauna, Z. E., Calcagno, A. M., et al. (2007) A “silent”
polymorphism in the MDR1 gene changes substrate specificity,
Science, 315, 525-528.
142.Zhang, G., Hubalewska, M., and Ignatova, Z.
(2009) Transient ribosomal attenuation coordinates protein synthesis
and co-translational folding, Nat. Struct. Mol. Biol.,
16, 274-280.
143.Zhou, M., Guo, J., Cha, J., Chae, M., Chen, S.,
et al. (2013) Non-optimal codon usage affects expression, structure and
function of clock protein FRQ, Nature, 495, 111-115.
144.Sander, I. M., Chaney, J. L., and Clark, P. L.
(2014) Expanding Anfinsen’s principle: contributions of
synonymous codon selection to rational protein design, J. Am. Chem.
Soc., 136, 858-861.
145.Hu, S., Wang, M., Cai, G., and He, M. (2013)
Genetic code-guided protein synthesis and folding in Escherichia
coli, J. Biol. Chem., 288, 30855-30861.
146.Kim, S. J., Yoon, J. S., Shishido, H., Yang,
Z., Rooney, L. A., et al. (2015) Protein folding. Translational tuning
optimizes nascent protein folding in cells, Science, 348,
444-448.
147.Yu, C. H., Dang, Y., Zhou, Z., Wu, C., Zhao,
F., et al. (2015) Codon usage influences the local rate of translation
elongation to regulate co-translational protein folding, Mol.
Cell, 59, 744-754.
148.Walsh, I. M., Bowman, M. A., Soto Santarriaga,
I. F., Rodriguez, A., and Clark, P. L. (2020) Synonymous codon
substitutions perturb cotranslational protein folding in vivo
and impair cell fitness, Proc. Natl. Acad. Sci. USA, 117,
3528-3534.
149.Komar, A. A. (2007) Silent SNPs: impact on gene
function and phenotype, Pharmacogenomics, 8,
1075-1080.
150.Sauna, Z. E., and Kimchi-Sarfaty, C. (2011)
Understanding the contribution of synonymous mutations to human
disease, Nat. Rev. Genet., 12, 683-691.
151.Hunt, R. C., Simhadri, V. L., Iandoli, M.,
Sauna, Z. E., and Kimchi-Sarfaty, C. (2014) Exposing synonymous
mutations, Trends Genet., 30, 308-321.
152.Simhadri, V. L., Hamasaki-Katagiri, N., Lin, B.
C., Hunt, R., Jha, S., et al. (2017) Single synonymous mutation in
factor IX alters protein properties and underlies haemophilia B, J.
Med. Genet., 54, 338-345.
153.Pechmann, S., Chartron, J. W., and Frydman, J.
(2014) Local slowdown of translation by nonoptimal codons promotes
nascent-chain recognition by SRP in vivo, Nat. Struct. Mol.
Biol., 21, 1100-1105.
154.Buhr, F., Jha, S., Thommen, M., Mittelstaet,
J., Kutz, F., et al. (2016) Synonymous codons direct cotranslational
folding toward different protein conformations, Mol. Cell,
61, 341-351.
155.Kimchi-Sarfaty, C., Schiller, T.,
Hamasaki-Katagiri, N., Khan, M. A., Yanover, C., and Sauna, Z. E.
(2013) Building better drugs: developing and regulating engineered
therapeutic proteins, Trends Pharmacol. Sci., 34,
534-548.
156.Komar, A. A. (2016) The art of gene redesign
and recombinant protein production: approaches and perspectives,
Top. Med. Chem., 2, 1-17.
157.Alexaki, A., Hettiarachchi, G. K., Athey, J.
C., Katneni, U. K., Simhadri, V., et al. (2019) Effects of codon
optimization on coagulation factor IX translation and structure:
implications for protein and gene therapies, Sci. Rep.,
9, 15449.
158.Hunt, R., Hettiarachchi, G., Katneni, U.,
Hernandez, N., Holcomb, D., et al. (2019) A single synonymous variant
(c.354G>A [p.P118P]) in ADAMTS13 confers enhanced specific activity,
Int. J. Mol. Sci., 20, 5734.
159.Knobe, K. E., Sjorin, E., and Ljung, R. C.
(2008) Why does the mutation G17736A/Val107Val (silent) in the
F9 gene cause mild haemophilia B in five Swedish families?
Haemophilia, 14, 723-728.
160.Gartner, J. J., Parker, S. C., Prickett, T. D.,
Dutton-Regester, K., Stitzel, M. L., et al. (2013) Whole-genome
sequencing identifies a recurrent functional synonymous mutation in
melanoma, Proc. Natl. Acad. Sci. USA, 110,
13481-13486.
161.Hamasaki-Katagiri, N., Salari, R., Wu, A., Qi,
Y., Schiller, T., et al. (2013) A gene-specific method for predicting
hemophilia-causing point mutations, J. Mol. Biol., 425,
4023-4033.
162.Hamasaki-Katagiri, N., Lin, B. C., Simon, J.,
Hunt, R. C., Schiller, T., et al. (2017) The importance of mRNA
structure in determining the pathogenicity of synonymous and
non-synonymous mutations in haemophilia, Haemophilia, 23,
e8-e17.
163.Katneni, U. K., Liss, A., Holcomb, D.,
Katagiri, N. H., Hunt, R., et al. (2019) Splicing dysregulation
contributes to the pathogenicity of several F9 exonic point
variants, Mol. Genet. Genomic Med., 7, e840.
164.Edwards, N. C., Hing, Z. A., Perry, A.,
Blaisdell, A., Kopelman, D. B., et al. (2012) Characterization of
coding synonymous and non-synonymous variants in ADAMTS13 using ex
vivo and in silico approaches, PLoS One, 7,
e38864.
165.Sauna, Z. E., Richards, S. M., Maillere, B.,
Jury, E. C., and Rosenberg, A. S. (2020) Editorial: immunogenicity of
proteins used as therapeutics, Front. Immunol., 11,
614856.