REVIEW: Superoxide Anion Radical Generation in Photosynthetic Electron Transport Chain
Marina A. Kozuleva1,a* and Boris N. Ivanov1
1Institute of Basic Biological Problems, Pushchino Scientific Center for Biological Research of the Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia* To whom correspondence should be addressed.
Received May 9, 2023; Revised June 16, 2023; Accepted June 18, 2023
This review analyzes data available in the literature on the rates, characteristics, and mechanisms of oxygen reduction to a superoxide anion radical at the sites of photosynthetic electron transport chain where this reduction has been established. The existing assumptions about the role of the components of these sites in this process are critically examined using thermodynamic approaches and results of the recent studies. The process of O2 reduction at the acceptor side of PSI, which is considered the main site of this process taking place in the photosynthetic chain, is described in detail. Evolution of photosynthetic apparatus in the context of controlling the leakage of electrons to O2 is explored. The reasons limiting application of the results obtained with the isolated segments of the photosynthetic chain to estimate the rates of O2 reduction at the corresponding sites in the intact thylakoid membrane are discussed.
KEY WORDS: photosynthesis, photosynthetic electron transport chain, oxygen reduction, superoxide radicalDOI: 10.1134/S0006297923080011
Abbreviations: Fd, ferredoxin; FNR, ferredoxin:NADP+ oxidoreductase; DCPIP, 2,6-dichlorophenolindophenol; DNP-INT, dinitrophenyl ether 2-iodine-4-nitrotimol; Em, midpoint redox potential; PETC, photosynthetic electron transport chain; PhQ, phylloquinone; PQ, plastoquinone; PSI, photosystem I; PSII, photosystem II; QA and QB, primary and secondary quinone acceptors of photosystem II, respectively; QO and QR, quinol-oxidizing (QO site) and quinol-reducing (QR site) sites of the b6f complex, respectively; ROS, reactive oxygen species.
INTRODUCTION
Molecular O2 is a byproduct of water oxidation in photosynthesizing organisms, which use it as a source of electrons for generating a reductant required in carbon metabolism reactions. At the same time, components of the photosynthetic apparatus in aerobic organisms can react with O2 molecules. In 1951, Alan Mehler discovered that illumination of thylakoids leads to hydrogen peroxide (H2O2) formation, and concluded that molecular O2 could serve as a direct electron acceptor from the reduced components of the photosynthetic electron transport chain (PETC) [1]. The process of electron transfer from PETC components to O2 molecules, accompanied by their reduction, is known as the Mehler reaction. The main function of PETC is to reduce NADP+, and vast majority of the research has been conducted to estimate the share of “non-productive” Mehler reaction in the overall electron flow in PETC under various operational conditions [2].
However, understanding oxidation processes of the PETC components by O2 molecules is crucial, not only for assessing the impact of Mehler reaction on effectiveness of CO2 fixation, but also for recognizing its role in facilitating this fixation. Synthesis of ATP, used in CO2 fixation reactions, is driven by the build-up of proton gradient across the thylakoid membrane. This gradient arises not only during linear electron transport to the oxidized pyridine nucleotide, but also during electron transport to oxygen as an acceptor (known as pseudocyclic electron transport), and during cyclic electron transport. The latter requires a certain necessary level of oxidation of the plastoquinone (PQ) pool (redox poising), which can be provided both by electron transfer to the acceptors of Photosystem I (PSI), particularly the Mehler reaction, or, possibly, by the direct oxidation of the PQ pool by oxygen (see below). The process of O2 reduction in PETC plays a critical role in maintaining homeostasis of the photosynthesizing cell and in adapting the entire photosynthetic organism to environmental conditions. This role is realized via formation of reactive oxygen species (ROS), such as the superoxide anion radical (O2•−), and H2O2. These ROS serve as primary signaling molecules that trigger adaptive metabolic readjustments. It is exactly the generation of ROS that allows PETC to function as a sensitive sensor of environmental changes, such as light intensity, temperature, water availability, soil salinity, and so on.
Unsurprisingly, many studies have been devoted to determining from which PETC components the transfer of electrons to O2 molecules is possible, and from which it predominantly occurs [3-7]. To date, new data have been accumulated about the mechanisms of O2 reduction in PETC, and new ideas have emerged regarding the conditions under which this process occurs and how the components of PETC that can be oxidized by oxygen have evolved. This review focuses on these new findings, analyzing earlier results in each case. Primary attention is given to the O2 reduction in PSI, which is generally considered as the main site in PETC where this process takes place.
CONDITIONS AND PATHWAYS OF O2 REDUCTION IN PETC
Twenty years after discovery of the Mehler reaction, it was demonstrated that it begins as a one-electron oxidation of PETC components by O2 molecules under illumination, resulting in formation of O2•− [8, 9]. From this point forward, the term “O2 photoreduction” is used synonymously with the term “formation of O2•− during the electron transfer from the PETC components to the O2 molecule”. When assessing thermodynamic feasibility of O2 reduction, it should be noted that PETC contains components dissolved in aqueous phase, membrane-associated components in contact with the aqueous phase, as well as components embedded in the hydrophobic zones of proteins and membrane. The midpoint redox potential, Em, for the O2/O2•− pair varies in different environments: –160 mV (measured against the normal hydrogen electrode, NHE) in water and approximately –550 to –600 mV in dimethylformamide, a model solvent for the membrane with dielectric constant of 36.7 [10]. The possibility of O2•− formation within the thylakoid membrane under the light was suggested in the works by K. Asada et al. [11], and experimentally confirmed in our studies with the EPR method, using the lipophilic cyclic hydroxylamine TMT-H when O2 was the only final acceptor [12, 13]. Subsequent research showed that light can induce formation of O2•− within the thylakoid membrane even when ferredoxin (Fd) and NADP+ were present. This suggests that the O2 reduction can occur simultaneously with the NADP+ photoreduction [14].
Under illumination, there may be more than one pathway for O2 reduction active in the PETC. The table presents the rates of O2•− formation reported in the literature at the main PETC sites: Photosystem II (PSII), PSI, cytochrome b6f complex, stromal pool of Fd, and membrane PQ pool. Properties and characteristics of each of the known O2 reduction pathways in the chloroplast PETC are discussed below.
Rates of O2•− generation in the various
segments of photosynthetic electron transport chain in higher
plants
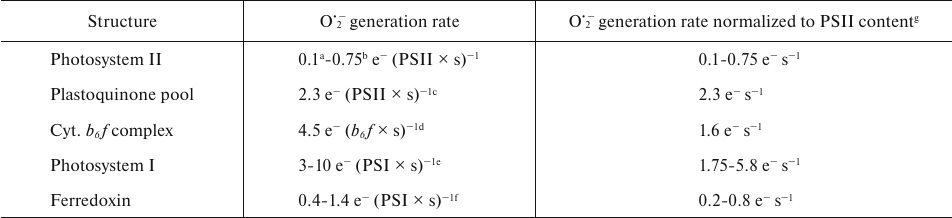
a Calculated from the cytochrome c reduction rate in
[15].
b Calculated from the O2 uptake rate in BBY
particles in [16], considering the Chlorophyll :
P680 ratio = 350 [17].
c Calculated from the diuron-independent O2 uptake
rate in thylakoids in [16], considering the
Chlorophyll : P680 ratio = 370 [18].
d From [19].
e From [20]; range of rates for
different light intensities (from lowest to highest intensity).
f Calculated, using the values of oxidation rate constant of
Fd by oxygen (0.08-0.28 s−1) [21,
22] and the ratio of Fd:PSI in chloroplasts = 5
[23].
g Calculated using the stoichiometry b6f :
PSII = 0.35 and PSI : PSII = 0.58 [24].
O2 reduction in Photosystem II. Many studies have been devoted to investigation of oxygen photoreduction in PSII; analysis of these studies is presented in the reviews [2, 4]. Most of these works have been carried out with PSII preparations of varying integrity: fragments of thylakoid membranes enriched with PSII (BBY-particles), and PSII complexes in which PQ is absent. To date, the studies appeared, which have attributed O2•− generating activity to PSII components not only in the isolated thylakoids but also in leaves. This has primarily been achieved by visualizing oxidized amino acid residues near the PSII cofactors [25-27] – an experimental approach based on the assumption that ROS produced by the electron transfer cofactors can first modify proximal residues in close proximity to the site of ROS generation [28]. However, this approach does not allow any quantitative evaluation of the rate of O2•− formation, whereas working with the PSII preparations isolated from thylakoid membranes makes it possible to measure the rate of light-induced O2•− generation. The rates for this process in the BBY-particles are given in the table.
Various components of PSII are considered as oxygen reductants. Formation of O2•− was registered in the D1/D2/cytochrome b559 complex, where quinones were absent at the QA and QB sites, suggesting that pheophytin, the primary electron acceptor in PSII, could reduce O2 [29]. Pheophytin, with the lowest Em value among the PSII cofactors (–610 mV), is capable of reducing O2 in the hydrophobic part of the protein, where it is located. In this region, potential of the O2/O2•− pair is close to this value, or even slightly more positive (see above). Moreover, it was recently shown that in the Arabidopsis vte1 mutant deficient in tocopherol biosynthesis, in which PSII lacks two tocopherol molecules characteristic of the wild type located near pheophytin and non-heme iron, generation of O2•− is increased and oxidized amino acid residues were detected near pheophytin [27]. The authors suggested that at high illumination, pheophytin produces O2•−, which in the wild type PSII oxidizes the adjacent tocopherol molecule rather than the surrounding amino acid residues. It is usually assumed that under moderate illumination, reduction of O2 by pheophytin is unlikely due to the short lifetime of its reduced form, 200-500 ps, during oxidation by the next electron carrier in PSII, PQ molecule at the QA site [2].
Reduction of O2 by the tightly bound PQ•− at the QA site has been suggested, in particular, based on the fact that low concentrations of the herbicide diuron, an inhibitor of electron transfer from QA to the next PETC component, the PQ molecule at the QB site, stimulated formation of O2•− in the pea thylakoids [30]. Generation of O2•− was recorded in the BBY complexes additionally treated to remove PQ molecules, thereby leaving the QB site vacant, using specific spin traps and reduction of exogenous cytochrome c [15]. Modified amino acid residues located closer to the QA site were also found in the spinach leaves and in the Arabidopsis vte1 mutant [25, 27].
The question of thermodynamics of the reaction between O2 and PQ•− at the QA site remains, however, debatable. It was shown that Em (QA/QA−) depends on the presence of a bicarbonate ion near the non-heme iron: –70 mV and –145 mV in the absence and presence of HCO3−, respectively [31]. The Em values of the pair (QA/QA•−) in the presence of HCO3− (–145 mV) and the pair O2/O2•− (–160 mV in water) are close, suggesting that from the thermodynamic point of view oxidation of PQ•− at the QA site by oxygen, although not beneficial, is plausible. The question is whether we can consider Em (O2/O2•−) in water, since QA is located in a rather hydrophobic part of PSII? This assumption is plausible, as QA interacts with water channels through which HCO3−, in particular, arrives at the non-heme iron [15, 32], and there are many polar and ionogenic amino acid residues in this region. Therefore, reduction of O2 by the tightly bound PQ•− at the QA site appears more favorable, when HCO3− is present on the acceptor side of PSII. However, under conditions where the stable PQ•− at the QA site induces release of HCO3−, the rate of O2•− production increases [15]. Based on this, it was concluded that the presence of HCO3− limits access of O2 to the QA site. The authors of the study [15] suggest that the molecule of O2 that is reduced to O2•− is bound to the non-heme iron, which increases the Em value of the pair O2/O2•−.
Reduction of O2 at the QB site is considered thermodynamically improbable due to the Em (PQ/PQ•−) at the QB site being +90 mV [33]. Indeed, even exposure of BBY particles to high light intensity causing photoinhibition did not result in appearance of the oxidized amino acid residues at the QB site [26]. It is likely that there is no generation of O2•− at this site.
Based on the coupling of O2 reduction with oxidation of cytochrome b559, which is part of the PSII complex and participates in the cyclic transport of electrons around PSII, it has been assumed in the literature that this cytochrome is responsible for O2 reduction [34, 35], when it is in a low potential form (Em = –40 – +80 mV [36]) or in a very low potential form (Em = –150 – –200 mV [37]). However, O2 reduction even by the low-potential forms of cytochrome b559 is thermodynamically unfavorable due to the cytochrome location in the hydrophobic zone, where Em value of the O2/O2•− pair is significantly lower than these values. It was suggested that either PQ•−, which is formed during the oxidation of PQH2 by cytochrome b559 at the PSII plastohydroquinone-binding QC site [38], or free PQ•−, which is produced in the comproportionation reaction (see below) of PQ with PQH2 formed at the QC site during oxidation of cytochrome b559 by the bound plastosemiquinone [2], act as the O2 reductant.
Thus, O2•− generation in PSII is possible via oxidation of pheophytin and PQ•− at the QA site (and possibly at the QC site). However, quantitative estimates of this process available in the literature, obtained for the BBY particles (0.1 e− (PSII × s)−1 [15], 0.25 e− (PSII × s)−1 [39], 0.75 e− (PSII × s)−1 [16]), indicate its low efficiency. Some estimates may even be exaggerated, as they were obtained for the BBY complexes, which retain 2 to 3 free PQ molecules per PSII reaction center, and these molecules, when reduced, can reduce O2•− (see below). Additionally, in some works [15, 39], experimental conditions prevented reliable quantitative estimation of O2•− generation due to the use of media with low pH values, at which the rate of spontaneous dismutation of O2•− is high, and it is almost impossible to achieve saturating concentrations of the trap for O2•−, that would ensure registration of all O2•− generated in the system [40]. In most experiments with PSII particles, O2 was the only electron acceptor from PSII cofactors, but even in this case, the rate of O2•− production was very low. Given the aforementioned evidence, it is unlikely that actual contribution of PSII to O2•− production in chloroplasts is significant.
O2 reduction in the plastoquinone pool of thylakoid membrane. The O2-dependent oxidation of the PQ pool, observed in darkness after illumination of thylakoids [41], suggested the possibility of electron transfer from the components of this pool to O2 molecules. Light-induced generation of O2•− was demonstrated in the isolated pea thylakoids in the presence of dinitrophenyl-2-iodo-4-nitrothymol (DNP-INT), a highly effective competitive inhibitor of PQH2 oxidation in the quinol-oxidizing site, Qo-site, of the b6f-complex [16, 30]. These conditions suggest that only PSII and components of the PQ pool can reduce O2. It was shown in [16] that the BBY-particles generated O2•− at a much slower rate than the thylakoids in the presence of DNP-INT (see table). This indicated that the light-induced generators of O2•− in thylakoids were molecules of the PQ pool. Based on the similarity of dependencies between the increased generation of O2•− in the PQ pool with increasing pH from 5.0 to 6.5 [16] and decrease in the redox potential differences between PQ/PQ•− pair and the O2/O2•− pair (in water) to negative values in this pH range, it was suggested [3, 16] that O2•− is formed in the reaction of O2 with the molecules of free PQ•− at the interface between the membrane and aqueous phase.
The source of free PQ•− in thylakoids under light could be, firstly, the comproportionation reaction PQH2 + PQ → 2PQ•− + 2H+. The steady-state concentration of PQ•−, produced in this reaction in the PQ pool, was calculated in [3]. It was in good agreement with the calculated concentration of PQ•− required to ensure the rates of O2•− production observed in [16]. It should be noted that in the case of PQ•− generation in the comproportionation reaction, the maximum rates of O2•− production in the PQ pool should be observed under conditions when the pool is half-reduced, while at high light intensities, when the PQ pool is almost fully reduced, the PQ•− content significantly decreases as a result of this reaction. Secondly, free PQ•− can also be formed as a result of PQH2 oxidation by hydrogen peroxide and superoxide radical, which are formed both in the PQ pool [16] and at other sites of the PETC, primarily in PSI at high light intensity [42]. Thirdly, another potential source of free PQ•− in the pool could be incomplete oxidation of PQH2 at the quinol-oxidizing site (Qo) of the b6f complex, followed by the release of PQ•− from this complex. The possibility of semiquinone leaving the quinol oxidation site in the bc1 complex was suggested in [43].
Considering the above, it can be assumed that quantitative estimates of the PQ pool contribution obtained using inhibitors of enzymatic oxidation of PQH2 do not completely reflect O2 reduction in the PQ pool in chloroplasts. Firstly, nearly complete reduction of the PQ pool in the presence of DNP-INT is observed at significantly lower light intensities than in the case with the operation of the full PETC [44]. Secondly, the use of DNP-INT or another inhibitor of PQH2 oxidation in the b6f complex blocks the electron flow in the chain and minimizes production of O2•− in the PSI, as well as in the b6f complex (see below). Thirdly, inhibitors at saturating concentrations prevent PQH2 oxidation at the Qo-site and exclude the third described above possible source of free PQ•−. Therefore, it seems likely that the maximum rates of O2•− production observed in the thylakoids treated with DNP-INT (see table) may be underestimated due to the use of inhibitors of enzymatic oxidation of PQH2. It is possible that the rates of O2•− production in the PQ pool of intact chloroplasts are higher than those reported in [16].
O2 reduction in the cytochrome b6f complex. Production of O2•− was demonstrated in the isolated b6f complexes (PQH2-plastocyanin oxidoreductase) to which reduced decylplastoquinone and plastocyanin were added as an electron donor and acceptor, respectively [19]. Simultaneously, reduction of plastocyanin was recorded. The authors hypothesized that the most probable source of O2•− in this system might be PQ•−, which forms at the Qo site after one-electron oxidation of PQH2 [19]. In this study, it was found that the rate of superoxide anion-radical production in the isolated b6f complex as a percentage of the electron transport rate was nearly an order of magnitude higher than in the isolated mitochondrial bc1 complex. It was estimated in another study [45] that at the Qo site of the b6f complex, Em (PQ/PQ•−) was quite low, –280 mV, and its reaction with O2 was thermodynamically possible.
First cofactor of the high-potential branch of the b6f complex cofactors, Fe2-S2 Rieske cluster, which receives the first electron during the incipient oxidation of PQH2 at the Qo site, has high Em (+330 mV) making its oxidation by O2 thermodynamically unfavorable. Participation of the low-potential heme of cytochrome b6 (b6L), the first cofactor of the low-potential branch of b6f complex cofactors, which receives the second electron during oxidation of PQH2 at the Qo site and has a rather negative Em, −150 mV [46] in the O2•− production was suggested [19]. Oxidized amino acid residues were found at the Qo site [28], indicating the possibility of O2•− production there. However, precise interpretation of the reactions leading to oxidative modifications is complicated, since the b6f complex contains a chlorophyll a molecule capable of producing 1O2 [47], as well as Fe2-S2 cluster of the Rieske protein, which, like other Fe-S clusters [48], may potentially catalyze production of HO• from H2O2 molecules. 1O2 and HO• have greater reactivity than O2•− and can modify amino acid residues more effectively than O2•−.
Role of ferredoxin in O2 reduction. The stromal protein Fd contains a single Fe2-S2 cluster and has low Em equal to −420 mV. In its reduced form it can effectively reduce O2 to O2•− in the aqueous phase. However, production of O2•− involving Fd occurs at low rates: the first-order rate constant for oxidation of the reduced Fd by molecular O2 is from 0.08 to 0.28 s−1 [21, 22, 49]. This is presumably due to the structure of its iron-sulfur active center; quinones with similar Em values of the Q/Q•− pair have O2 reduction rate constants about 6 orders of magnitude higher [10]. Considering the values of the rate constants of the reaction and the ratio of Fd : PSI in the chloroplasts of higher plants [23], the rate of Fd-dependent photoreduction of O2 in the chloroplast does not exceed 10% of the maximum rate of O2 photoreduction in PSI (table).
Nevertheless, Fd was long considered as a primary participant in O2 photoreduction in chloroplasts [50] based on the frequently observed significant stimulation of O2 uptake and O2•− production when Fd was added to the isolated thylakoids of spinach/pea/Arabidopsis deprived of stromal components during extraction [14, 51, 52]. In such experiments, the ratio of Fd to PSI was three orders of magnitude higher than in vivo. Due to the slow oxidation of reduced Fd, it led to significant accumulation of the reduced Fd ensuring the observed high rate of O2 reduction. Addition of NADP+, main electron acceptor from the reduced Fd, significantly decreased contribution of Fd to O2•− production by the thylakoids [14, 53]. Apparently, efficiency of NADP+ regeneration in the Calvin–Benson–Bassham cycle determines contribution of Fd to O2•− production in vivo, due to the change in the number of reduced Fd molecules accessible to oxidation by oxygen.
In the literature, attempts have been made to assess involvement of Fd in the reduction of O2 in chloroplasts (i.e., at the native Fd : PSI ratio). It was done by comparing Michaelis constants, Km(O2), measured for the Mehler reaction in the isolated thylakoids and in the intact chloroplasts/cells/leaves. This approach has been thoroughly described in [54], and is based on the assumption that the higher Km(O2) in more complex structures reflects involvement of multiple sites in O2 photoreduction. The Km(O2) values for chloroplasts and whole cells (50-95 µM) were one order of magnitude higher than the Km(O2) for thylakoids (3-10 µM). An obvious conclusion drawn from such comparison was that Fd serves as an additional site in more complex structures. However, the situation is more complex than it seems at first glance.
Firstly, there are questions about the Km(O2) values used when comparing different structures. The assumption that PSI is the only site of O2 photoreduction in the conducted experiments with isolated thylakoids is likely not entirely accurate. The Km(O2) value for O2 photoreduction in PSI, 3 µM, obtained in one of the studies [55] was evidently underestimated due to the use of 2,6-dichlorophenolindophenol (DCPIP) as an artificial electron donor to PSI [56] (see more details below). On the other hand, reliability of distinguishing the Mehler reaction and other O2-consuming reactions in more complex structures including mitochondrial respiration, Rubisco oxygenase reaction (photorespiration), oxidation of the PQ-pool by plastid terminal oxidase (chlororespiration), O2 uptake due to the production of 1O2 and peroxidation of lipids, reduction of O2 to water involving proteins containing two iron atoms in contact with the flavin group (flavodiiron proteins, Flvs or FDPs), which are absent in angiosperms, but present in cyanobacteria, green algae and other higher plants is also questionable. Thus, depending on the organism studied and conditions in which the measurements were carried out, the obtained Km(O2) value may be associated not only with the Mehler reaction.
Secondly, in addition to the chloroplast stromal Fd, other stromal components may be involved in the direct photoreduction of O2 in vivo. In the literature, there have been suggestions about the roles of nitrite reductase, Fd-reducible glutamate synthase [57], and monodehydroascorbate reductase [58] in this process. Contribution of these proteins to the light-induced O2 reduction may affect the measured value of Km(O2) for the intact systems, but their contribution to this process has not yet been determined due to the fact that the mentioned studies were not continued. Most likely, these enzymes could participate in O2 reduction only under conditions of deficiency of their specific substrates [57].
Therefore, comparing Km(O2) values for different structures with the aim of determining contribution of Fd to the reduction of O2 in chloroplasts represents an approach that makes it difficult to draw reliable conclusions. Moreover, quantitative assessment of O2 photoreduction pathways in the suspension of pea thylakoids in the presence of Fd showed that the increase in Fd concentration stimulated not only reduction of O2 involving the reduced Fd, but also O2 reduction by the membrane components [49, 53]. To explain the latter effect, it was hypothesized that the increase in the electron flow from PSI to Fd with the increase in its concentration might alter the ratio of direct electron transport pathways and charge recombination in PSI in such a way that concentrations of the reduced forms of intermediate acceptors of this photosystem, phylloquinone in A1 site and iron-sulfur center FX, increases, and the electron flow from them to O2 increases also [53]. An alternative assumption could be that Fd initiates or stimulates a certain pathway of O2 photoreduction in thylakoids, which is inactive or minimally active in the absence of Fd. For example, such pathway could be O2 reduction involving the membrane-bound FNR (ferredoxin:NADP+ oxidoreductase), which receives electrons from PSI only in the presence of Fd. It is known that the exogenous addition of FNR to thylakoids highly stimulates O2 reduction [14, 58]. However, recent experimental results argue against significant participation of FNR in O2 reduction in thylakoids [14]. In this study, production of the membrane-bound O2•− was measured in thylakoids from Arabidopsis, isolated from both wild-type plants and a mutant deficient in the FNR1 isoform [59]. This mutant is characterized by the absence of FNR in isolated thylakoids [60]. It turned out that the rates of O2•− production in the membrane, both in the presence and absence of Fd, were the same in both genotypes, ruling out direct involvement of FNR in the production of O2•− in the thylakoids from the wild-type plant.
Another site of O2 reduction, which receives additional electrons in the presence of Fd, could be the cytochrome b6f complex. Some authors consider this complex to be a Fd-PQ oxidoreductase that participates in the cyclic electron transport around PSI [61, 62]. Based on this model, Fd donates one electron to reduce PQ at the quinone-reducing site (QR) of the complex, while the second electron comes from the Qo site. If this pathway is operating, it is possible that the presence of an electron flow from Fd to the b6f complex could influence lifetime of PQ•− at the Qo site and probability of its reaction with O2 (see above).
O2 reduction in Photosystem I. This photosystem has long been recognized as the principal site of O2•− generation during the Mehler reaction (see references in [63]), and indeed, among all PETC components, it is characterized by the highest rates of O2 photoreduction (table). However, contradictory assessments of its activity are reported in the literature. This is most pronounced when the rate constants of the O2 reduction reaction in PSI (k2), published in various studies are compared. The range of k2 values available in the literature is anomalously broad: from 7 × 102 M−1 s−1 to 107 M−1 s−1.
The chronological first estimation of k2 (107 M−1 s−1) was obtained for the spinach thylakoids, in which PSI functioned in isolation (i.e., diuron was added to inhibit PSII activity, and artificial electron donors were added to reduce P700+) [55]. However, this estimation is close to the rate constant for the reduction of methyl viologen by the terminal cofactors of PSI (1.5 × 107 M−1 s−1; [64]), which suggests similar efficiency of O2 and methyl viologen as immediate electron acceptors from PSI. This is unlikely, since methyl viologen significantly enhances the electron flow “through” PSI [42, 56]. The value of 107 M−1 s−1 is likely an overestimation, probably due to the use of reduced DCPIP as an electron donor for P700+ in the cited study [55]. It has been demonstrated with the isolated PSI complexes from Synechocystis and pea thylakoids that DCPIP acts as a redox mediator between PSI and O2, similar to methyl viologen. That is, the singly reduced form of DCPIP on the acceptor side of PSI is effectively oxidized by oxygen [56, 65]. Therefore, estimations of k2 and other characteristics of the PSI reaction with O2 (for example, Km(O2)) using DCPIP are erroneous, as they reflect the sum of reactions of O2 photoreduction by PSI cofactors and reduced DCPIP.
The lowest values for k2, 7.2 × 102 and 6.1 × 103 M−1 s−1, presented in the study [66], were calculated from the experimental data with pea thylakoids [42]. However, the O2 reduction rates in [42] were measured at atmospheric O2 content, which is a saturating concentration for the reaction of O2 reduction in PSI, whereas in order to correctly estimate the rate constant of the reaction, the use of the substrate at rate-limiting concentrations is required, i.e., in this case, when the oxygen reduction rate is dependent on its concentration. This is exactly how the measurements were conducted in the studies [20, 55, 67].
The most recently reported k2 values, ranging from 0.6 × 105 to 3.7 × 105 M−1 s−1 (depending on light intensity; see below), were obtained using the natural electron donor for PSI, plastocyanin [20]. Moreover, these values were also obtained in the presence of Fd, FNR, and NADP+, when the terminal acceptors of PSI reduced O2 concurrently with photoreduction of Fd with subsequent electron flow to NADP+, i.e., under conditions close to physiological. In the absence of Fd, the k2 values did not change significantly. Thus, the k2 values as well as the rate of O2 reduction measured at atmospheric O2 content presented in the study [20] are the closest characteristics of O2 reduction in PSI in vivo.
Which PSI cofactors can reduce O2? For a long time, it has been believed that electrons are transferred to O2 from the terminal cofactors of PSI, Fe4-S4 clusters FA/FB, located at the PsaC subunit on the stromal side of the PSI complex [9, 64]. Later, it became clear that the intermediate cofactors of PSI electron flow also contribute to O2•− production. It was, in particular, demonstrated in the study of light-induced H2O2-dependent iodination of the thylakoid membrane proteins that during the first seconds of illumination, O2 reduction is carried out by the cofactors situated at the PsaA and PsaB proteins, while longer illumination leads to the appearance of H2O2 in other parts of the thylakoids, including the protein area near FA/FB [11]. The authors suggested that O2 is reduced by the cluster preceding the FA/FB clusters in the electron flow chain, the FX cluster, located between the PsaA and PsaB subunits.
A hypothesis has been proposed regarding participation of the phylloquinone molecules, PhQ [68], a secondary cofactor of electron transfer, in O2 reduction. These are located in the A1 sites of the two pseudo-symmetrical branches of cofactors in PSI, A and B, and precede the FX cluster. In that study, thylakoid membranes were treated with hexane, which led to extraction of all PQ pool molecules and one PhQ molecule from the PSI located in the A branch, PhQA. Such membranes did not demonstrate O2 uptake in response to light flashes. Addition of PhQ in the form of vitamin K led to reappearance of O2 uptake, but only in response to the first light flash. The authors suggested that the hexane treatment modified the A1 site in such a way that its affinity for PhQ decreased [68].
Reduction of O2 with participation of PhQ in the native PSI complexes under steady-state illumination was first studied using complexes isolated from the cyanobacterium Synechocystis sp. PCC 6803 [69], assuming that both the composition of PSI electron transfer cofactors and amino acid environment in the A1 site are relatively conserved among cyanobacteria, algae, and higher plants. The study used a wild type strain and a strain with blocked PhQ biosynthesis (menB mutation). It was previously shown that PQ molecules were incorporated into the A1 sites of the mutant causing increase in Em (Q/Q•−) by ~100 mV relative to Em in the wild type strain. It also led to a 1000-fold increase in the lifetime of semiquinone in both branches [70]. The PSI complexes from the mutant showed significantly lower rates of O2 photoreduction compared to the complexes from the wild type strain [69], which was explained by the greater ability of PhQ•− in the A1 sites of the wild type to reduce O2 compared to PQ•− in the mutant sites.
Contribution of the individual PSI electron transport cofactors was revealed through investigation of the influence of light intensity on the k2 value for the PSI complex, isolated from the unicellular alga Chlamydomonas reinhardtii [20]. Increase in the apparent value of k2 with the increased light intensity was observed, and this was interpreted as evidence of several O2 photoreduction sites operating in PSI, each characterized by its own rate constant for this process and achieving maximum efficiency at the specific for this site light intensity. Experimental analysis using methyl viologen, a highly effective acceptor of electrons from the terminal cofactors of PSI, demonstrated that involvement of the terminal cofactors FA/FB in O2 reduction reaches its maximum at low light intensity, at which the FA/FB clusters are saturated with electrons. The apparent k2 increase with the increased light intensity is likely associated with the increase in contribution to O2 reduction from the preceding electron transport cofactors in PSI, when they are saturated with electrons. The roles of FX and PhQ were elucidated using sequential removal of Fe4-S4 clusters through specific treatments: the removal of FA/FB led to a slight decrease in the rate of O2 reduction across a wide range of light intensities, while additional removal of FX, resulting in PhQ in the A1 sites becoming the terminal cofactor, led to significant stimulation of O2 reduction. The latter is in agreement with the hypothesis of PhQ playing a key role in O2 reduction. PSI complexes, isolated from the mutant PsaA-F689N C. reinhardtii, in which Phe at position 689 of the PsaA protein was replaced by Asn, thereby increasing lifetime of PhQA•− from 0.25 µs to 17 µs [71], exhibited much higher rates of O2 photoreduction across a wide range of light intensities [20]. These data also indicate increase in the contribution of PhQ•− to the generation of O2•− in PSI with increasing light intensity.
In the isolated PSI complexes, two O2 reduction sites are active: the terminal FA/FB clusters and PhQ in the form of semiquinones at A1 sites. Contribution of each site depends on the conditions. At low light intensity, the rate of O2 photoreduction decreases in the presence of Fd, FNR, and NADP+, as the electron flow from PSI diminishes accumulation of electrons on the FA/FB clusters [20]. At the same time, presence of Fd, FNR, and NADP+ did not suppress O2 photoreduction observed at high light intensity indicating that PhQ is responsible for O2 reduction under conditions of parallel electron transport to NADP+ in this circumstance.
Surprisingly, the approach based on detection of the oxidized amino acid residues, which was successfully applied to determine the O2•−-generating activity of PSII and b6f complex cofactors (see above), proved to be unsuitable for visualizing O2•− production in PSI. The oxidized residues were not found in close proximity of the FA/FB clusters in spinach PSI complexes grown under field conditions [72]. Also, no modified residues were found in the immediate vicinity of the FX cluster and PhQA [72]. Conversely, two modified residues were detected near the PhQ in the B-branch (PhQB). However, interpretation of these results is complicated due to location of the chlorin ring in the chlorophyll a molecule between PhQB and these residues [72]. It could be hypothesized that the O2•− produced by the FA/FB clusters easily diffuses from the PsaC protein into the stroma (the FB cluster is located 3-4 Å from the surface of PsaC) and does not modify amino acid residues. Possibly, O2•− from the A1 sites also efficiently diffuses to the stromal side of the membrane, not reacting with the adjacent amino acid residues, since existence of the water-filled cavities leading from the A1 sites was demonstrated for PSI from the cyanobacterium Synechocystis sp. PCC 6803 [73].
It should be noted that the rates of O2•− generation in the isolated PSI complexes (table) may not fully reflect actual O2•−-generating activity of PSI in thylakoids and chloroplasts. O2 reduction in the isolated PSI complexes from Synechocystis [69] and C. reinhardtii [20] did not reach saturation with increasing light intensity across a wide range of light intensities (up to 2000 µmol photons m−2 s−1), while O2 reduction by the isolated, functionally active PSI in higher plant thylakoids (in the presence of diuron and artificial electron donors) tended to saturate at 500-600 µmol photons m−2 s−1 [42, 65]. On the one hand, this discrepancy might be due to introduction of the redox mediators such as N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) and sodium ascorbate to maintain PSI activity in the thylakoids. The oxidized forms of these compounds could accept electrons from the terminal cofactors of PSI [74-76], which should reduce accumulation of electrons on the PSI cofactors. On the other hand, the isolated PSI complexes might lack certain regulatory components that determine O2•− generating activity of PSI in the thylakoids. In particular, existence of a protein that regulates O2 reduction in PSI was postulated based on the comparison of the effects of short-day and long-day conditions on the Mehler reaction in tobacco plants [77]. It turned out that the short-day conditions favor higher rate of the PSI-dependent photoreduction of O2 in thylakoids and leaves. The authors suggested that a certain protein binds to PSI under the short-day conditions and facilitates diffusion of O2 to the site of its photoreduction, stimulating generation of O2•−. It has also been shown that the PsaE subunit, which together with the PsaC and PsaD subunits forms a docking site for Fd, could determine the degree of electron leakage to O2 [78].
The study [77] suggested that FNR might be the hypothetical protein regulating photoreduction of O2 in PSI. It has been shown that FNR and PSI, isolated from C. reinhardtii, interact with each other in a 1 : 1 stoichiometry, particularly involving the PsaE subunit [79], and the authors suggested that FNR might function as a subunit of PSI. FNR binding to PSI via the PsaE subunit was also demonstrated for barley [80]. In higher plants, the main proteins binding FNR are TROL and Tic62 [81, 82]. However, if the interaction with Tic62 and TROL is disrupted, FNR can interact with alternative, weaker binding sites on the thylakoid membrane, including PSI [60]. It is possible that the attachment of FNR to PSI could affect diffusion of O2 to the cofactors of PSI and/or initiate redistribution of the O2 reduction pathways within PSI.
EVOLUTIONARY ASPECTS
Effective electron transfer from PETC components to O2 leads to excessive production of such reactive oxygen species as O2•−, H2O2, HO•, and also reduces quantum yield of the light reactions of photosynthesis. The PETC of oxygenic phototrophs evolved into its current form under conditions of ongoing O2 production in light. It is logical to assume that minimizing the reactions of PETC components with O2 was one of the directions in the evolution of photosynthetic apparatus. Evolution of various photosynthetic complexes as they adapted to an emergence of oxygen-rich atmosphere has been addressed in several recent reviews [6, 83-86]. These studies particularly emphasize evolutionary strategies that the photosynthetic apparatus might have developed to minimize formation of singlet oxygen (1O2), which definitely was among the evolutionary trajectories of these systems. In this review, we considered changes that could have occurred in the photosynthetic apparatus to minimize non-productive electron leakage to O2 and excessive O2•− production.
In our opinion, evolutionary changes should first affect the pools of mobile carriers, as their reduced state is necessary for productive electron transport. The Em values of plastocyanin and cytochrome c6, electron donors to PSI, are high enough to exclude the possibility of their reaction with O2. However, Fd and the components of the PQ-pool, even in the modern PETCs possess low enough potentials for O2•− generation, which has been experimentally observed (see above).
Final product of the light stages of photosynthesis, reduced Fd, serves as an electron donor not only for NADP+ reduction but also for other metabolic pathways in the chloroplast [87]. To implement these pathways, the reduced Fd must diffuse in the stroma of the chloroplast. Obviously, this requires low efficiency of the Fd reaction with O2. In modern phototrophs with oxygenic photosynthesis, prosthetic group of Fd is the Fe2-S2 cluster, which is sufficiently deeply embedded in the protein. In modern phototrophs with anoxygenic photosynthesis, which have type I reaction centers, the role of electron acceptor is performed by Fd with two Fe4-S4 clusters, one of which is located almost on the surface of the protein [83], making the cluster accessible to O2 molecules. It has been suggested [83, 84] that embedding the cluster deeply within the protein restricts access of O2 molecules to it and reduces the likelihood of electron transfer to O2. Thus, replacement of the dicluster Fe4-S4·Fd with a monocluster Fe2-S2·Fd, with a relatively deeply embedded cluster in the protein, during the evolution of phototrophs could have been driven by adaptation to functioning in the presence of O2.
In the modern PETC, dicluster Fd has been preserved in the form of the PsaC subunit carrying two Fe4-S4 clusters in the form of cofactors of PSI, intermediate FA and terminal FB, which reduce the mobile monocluster Fd. Rapid efflux of electrons from FB to O2 is undesirable, as it could reduce efficiency of Fd reduction, which depends on the diffusion exchange of the reduced Fd for the oxidized Fd on the acceptor side of PSI. A higher Em (FA/FA−) compared to Em (FB/FB−) ensures longer residence time of the electron on FA than on FB [88, 89]. Thus, inversion of the redox potentials of FA and FB could be an evolutionary adaptation to minimize reduction of O2 by the FB cofactor in the absence of Fd [83, 84]. The FA cofactor is embedded in the protein and located in an area with sufficiently low dielectric permeability [90]. Raising Em (FA/FA−) also reduces thermodynamic probability of its reaction with O2.
In the energy-converting membranes of the very first organisms, menaquinone likely functioned as a liposoluble mobile carrier of protons and electrons, which to this day is present in a number of anaerobic phototrophs [91]. Redox potentials of the pairs (Q/Q•−) and (Q/QH2) of menaquinone are approximately 100 and 180 mV lower, respectively, than the corresponding pairs of PQ, i.e., the reduced forms of menaquinone are significantly easier can be oxidized by O2 than the forms of PQ. Therefore, in the presence of O2, menaquinones are less efficient as electron carriers from the type II reaction centers to the bc-type cytochrome complexes than such high-potential quinones as PQ or ubiquinone. Thus, the evolutionary replacement of menaquinone with PQ, a quinone with a more positive Em value, in the membranes of organisms with an oxygenic type of photosynthesis appears to be an important step in optimizing photosynthetic apparatus to the conditions of an oxygen atmosphere.
However, PQ•− has an Em allowing it to reduce O2 molecules to form O2•− in aqueous environment (see above), albeit at low rates. Semiquinones generally react with O2 molecules in a kinetically efficient manner [10]. In the PETC, there are several sites where PQ is consecutively reduced to PQH2, and where PQH2 is oxidized to PQ, forming in both cases an intermediate semiquinone form. Apart from PQ•−, the semiquinone form of the isoalloxazine portion of FAD is also transiently formed in FNR during sequential oxidation of two molecules of Fd and during reduction of NADP+. An effective oxidation of these cofactor semiquinone forms by oxygen at the moment when they should receive or give the second electron could disrupt normal electron transfer. Therefore, in our view, the next global trend in adaptation of photosynthetic apparatus to operate in the presence of O2 was to prevent O2 reactions with the intermediate semiquinone forms of PETC components during their two-step reduction or oxidation.
The semiquinone form of FAD reacts with O2 with high efficiency [92]. Possible mechanism for preventing oxidation of the semiquinone form of FAD by oxygen is discussed in the review [85]. It has been noted that the FNR of the phototrophs with oxygenic type of photosynthesis has two orders of magnitude greater catalytic activity than the FNR of anaerobic organisms [93], even though affinity of FNR for Fd may be similar [94]. High catalytic activity is achieved due to conformational changes induced by NADP+ binding, which significantly enhance both oxidation of Fd [95] and dissociation of the oxidized Fd molecules from the complex with FNR [96]. Such increase in the FNR catalytic activity likely reduces the probability of both oxidation of the semiquinone form of FAD by oxygen and formation of the Fd/FNR− complex in the absence of NADP+.
PQ•− is formed during the single reduction of PQ at the QB site of PSII and the QR site of the b6f complex. Apparently, at the QB site of PSII, the issue of decreasing electron leakage to O2 molecules is addressed on a thermodynamic level due to the high Em (QB/QB•−), +90 mV [33], as a result of which the QB•− reaction with O2 is thermodynamically unfavorable even in aqueous phase. No PQ•− generation has been demonstrated at the QR site of the b6f complex, although semiquinone appearance was recorded at the similar site of the bc1 complexes of purple bacteria [97] and mitochondria [98]. One of the key differences between the bc1 and b6f complexes is the presence of an additional heme in the cytochrome b6 – covalently linked heme cn. It has been suggested that, as part of the Q-cycle operation, the first of two electrons needed to reduce PQ at the QR site of the b6f complex is transferred from the high-potential heme of the cytochrome b6 (b6H) to the heme cn [99], and from the heme cn, an electron is transferred to the PQ molecule only simultaneously with the transfer of the second electron from the heme b6H [7]. Such mechanism minimizes lifetime of PQ•− at the QR site and, accordingly, reduces the probability of its reaction with O2.
At the Qo site of the b6f complex, it is believed that PQH2 is sequentially oxidized to PQ through a concerted mechanism, i.e., successive acts of electron transfer to the Fe2-S2 Rieske center and to the low-potential heme b6L. Efficient operation of the Q-cycle ensures efficient outflow of electrons from PQ•− at the Qo site along the low-potential branch of b6f complex cofactors. However, if there are few oxidized PQ molecules in the PQ pool, or if the heme b6L in the b6f complex is already reduced, a reverse electron transfer could occur from the heme b6L (Em (b6L/b6L−) = –150 mV) to PQ•− (Em (PQ•−/PQH2) = +480 mV) [7]. This reduces the probability of reaction between O2 and either heme b6L, or PQ•− at the Qo site. Disruption of the concerted oxidation could also occur under conditions of photosynthetic control, when the proton release from the Qo site to the lumen slows down, and PQH• cannot be deprotonated by the amino acid residue Glu78 of the subunit IV, which remains in a protonated state, as was recently hypothesized [100]. Since Em (PQ/PQH•) is higher than Em (PQ/PQ•−), electron leakage from the protonated plastosemiquinone to O2 is less probable. We suggest that the retardation of proton removal from the Qo site and increase in the lifetime of PQH• by maintaining Glu78 in a protonated state comprise an important mechanism for preventing the PQ•− reaction with O2 at the Qo site.
Replacing of the free menaquinone in the membrane pool with the higher potential quinone resulted in the increase of Em (by about 110-150 mV) of all tightly bound cofactors in the partner proteins of the mobile membrane carrier pool, i.e., in the type II reaction center and cytochrome bc1 complex [101, 102]. This, in turn, led to the decrease in the probability of these cofactors being oxidized by O2 molecules. Apparently, increase in Em was enough to solve the problem of electron leakage to O2 in the cytochrome b6f complex. Among all its tightly bound cofactors, heme b6L has the lowest Em value, –150 mV [103], which is insufficient to reduce O2 in the hydrophobic regions of the membrane. Presence of an electron on the heme cn should support efficient outflow of electrons from the heme b6L (see above) minimizing its reactions with O2.
However, for PSII, increasing Em may not be enough to minimize oxidation of its cofactors by oxygen. The tightly bound PQ at the QA site under normal operating conditions is singly reduced. Efficiency of the productive electron transfer from PQ•− at the QA site to PQ at the QB site is determined by the presence of HCO3− near the non-heme iron, which reduces Em (QA/QA−) from –70 mV to –145 mV as shown in the study [31]. However, it has been shown in another work that HCO3− on the acceptor side of PSII blocks the potential channel through which O2 molecules could diffuse to the QA site, thereby limiting access of O2 molecules to the site [15], which reduces formation of O2•−.
Pheophytin has a sufficiently low Em (–600 mV) to reduce O2 even in hydrophobic parts of the protein. However, suppression of the non-productive electron leakage to O2 is achieved at the kinetic level: short lifetime of the reduced pheophytin due to electron transfer to QA (200-500 ps) and recombination of the reduced pheophytin with P680+ (4-30 ns), apparently, significantly reduces the possibility of this cofactor reacting with O2.
In the type I reaction centers of modern anaerobic organisms, there are no tightly bound quinones [104-106] and, in heliobacteria in particular, menaquinones function as a mobile, lipid-soluble electron acceptors, an alternative to ferredoxin [107]. Presence of two acceptor pools could be advantageous in terms of the efficiency of photosynthetic reactions and protection of the photosynthetic apparatus from excessive illumination. In PSI (a type I reaction center, inherent exclusively to phototrophs with oxygenic photosynthesis) menaquinone (or more accurately, in most organisms, its derivative – PhQ) remains a tightly bound cofactor serving as a single-electron carrier, i.e., it does not get reduced to hydroquinone [108]. Unlike electron transfer between QA and QB in PSII, stoichiometry of which is 1 : 1, in PSI, the quinones of the two A1 sites transfer an electron to one Fe4-S4 cluster FX. Under conditions of increased illumination and limited electron flow from the stromal acceptors of PSI, a situation may occur when the iron-sulfur clusters of PSI will be predominantly reduced, and the two A1 sites will compete for one FX cluster. In this case, there may be a risk of charge recombination of quinones with P700+, including by a mechanism leading to formation of 3P700, as a result of which the risk of generating 1O2 [84] would increase. Oxidation of PhQ•− by O2 in this case would be a potentially less dangerous process, decreasing over-reduction of the ETC and lowering the risks of generating 1O2. Similar processes could occur in the QA site in PSII. However, given that Em (PhQ/PhQ•−) is much more negative than Em (PQ/PQ•−), it is significantly more challenging to minimize electron leakage from PhQ•− to O2 compared to PQ•−. At the same time, maintaining low Em values for PSI cofactors is necessary to reduce such a low-potential electron carrier as ferredoxin.
Kinetic control (i.e., rapid electron transfer to the next cofactor in the chain) likely also cannot be fully implemented in PSI, where two PhQ are present, differing in Em by 170 mV [90] and in the lifetime of the semiquinone form by one order of magnitude [109]. Two PhQ under steady-state illumination conditions could compete for one FX cluster, which increases probability of the electron leakage to O2 from the longer-lived PhQ•− in the A-branch. Limiting of the O2 accessibility to PhQ in the A1 sites seems to be only marginally feasible evolutionary strategy. Unlike PSII, PSI does not have channels for either influx/efflux of the reaction substrates/products, or for bicarbonate ions. In the study modeling the structure of cyanobacteria PSI, presence of aqueous cavities connecting the A1 sites to the acceptor side has been suggested [73] through which not only O2 molecules are assumed to diffuse, but also much larger molecules such as methyl viologen.
Thus, we suggest that the reaction of O2 with PhQ in the A1 sites could not have been minimized sufficiently, and to this day, among all components of the PETC, the highest rates of O2•− production are characteristic for PSI due to the presence of PhQ in it. Under conditions of over-reduction of the chain, when the intensity of light exceeds the capabilities of metabolic utilization of light energy, O2 becomes an available additional electron acceptor, capable of sustaining electron transport and minimizing over-reduction of the PETC components, thereby mitigating photoinhibition. Therefore, it is likely that evolution of the photosynthetic apparatus proceeded not merely towards minimizing electron transfer to O2, but towards regulating this process.
CONCLUSION
The above analysis considered potential mechanisms for the reduction of O2 to O2•− by various components of the PETC, as well as evolutionary transformations that the PETC might have undergone to reduce non-productive electron leakage to O2. The table summarizes estimates of O2•− generation rates at different segments of the PETC available in the literature. The table also includes rates normalized to the content of PSII, allowing comparison of the potential contributions of different components to the production of O2•− in chloroplasts.
However, such comparison requires consideration of the conditions under which these rates were measured. Firstly, the rates presented in the table were obtained using various experimental approaches under different conditions and for different organisms. Secondly, many of these rates were obtained for the isolated structures (for PSII, PSI, b6f-complex) or in the presence of inhibitors (for the PQ-pool). Consequently, interrelations between the PETC components could have been altered and some components that are present in whole membranes might have been absent. Thirdly, in some cases (PSII, PQ-pool), O2 served as the only available electron acceptor. It is difficult to definitively assess how this could affect the probability of O2•− generation in these cases. For example, presence of NADP+ reduces O2•− production by the reduced Fd [53], whereas presence of Fd and NADP+ does not significantly affect O2•− production in PSI [20]. It should also be noted that normalization presented in the table is based on the relative content of the individual pigment–protein complexes in a specific organism (Arabidopsis thaliana) under specific conditions [24]. The content of PSII, b6f-complex, PSI, and relative size of the photoactive PQ-pool, and their ratio, vary in plants depending on environmental conditions [110, 111], and relative contribution of these components to O2•− production, evidently, could also vary.
Despite these limitations, comparison of the rates presented in the table provides a useful model and approximation for the comprehensive description of O2 reduction in PETC. It is clear that contribution of PSII is the smallest, but even Fd, previously considered as the primary reducer of O2 (see above), generates O2•− at approximately the same rates as PSII. It should be noted that calculation of rates for Fd is based on the assumption that the reduced Fd is accumulated in the chloroplasts, and therefore the rates are close to maximum. The PQ-pool and the cytochrome b6f-complex contribute nearly equally to the total rate of O2 photoreduction in chloroplasts. Contribution of PSI depends on light intensity, and under high light intensity, PSI produces approximately half of all O2•− generated in this PETC model.
PSI is commonly regarded as the main site of O2 reduction in the PETC. This notion is supported by several indirect pieces of evidence (though these could also be interpreted in favor of other segments of PETC) and by a number of experimental results based on the use of mutants or inhibitors, which also allows for ambiguous interpretation. Our analysis shows that no single component of the PETC can generate O2•− as effectively as PSI under high light intensity. But from these data, it is also clear that the components of the PETC without PSI can collectively generate O2•− at the rate comparable to that in PSI. Therefore, while it is reasonable to consider PSI the place where O2•− is produced at the highest rates under illumination, it may not be appropriate to consider PSI as the dominant site of its production under all circumstances.
Contributions. M.A.K. – writing the text; B.N.I. – editing the article.
Funding. This work was financially supported by the Russian Science Foundation, grant no. 22-24-01074.
Acknowledgments. The authors would like to thank Dr. M. M. Borisova for valuable discussions during preparation of the review.
Ethics declarations. The authors declare no conflict of interest in financial or any other sphere. This article does not contain descriptions of any research involving human or animal subjects performed by any of the authors.
REFERENCES
1.Mehler, A. H. (1951) Studies on reactions of
illuminated chloroplasts: I. Mechanism of the reduction of oxygen and
other hill reagents, Arch. Biochem. Biophys., 33, 65-77,
doi: 10.1016/0003-9861(51)90082-3.
2.Ivanov, B. N., Khorobrykh, S. A., Kozuleva, M. A.,
and Borisova-Mubarakshina, M. M. (2013) The role of oxygen and its
reactive species in photosynthesis, in Photosynthesis: Questions to
Answer and What We Know Today (Allakhverdiev, S. I., Rubin, A. B.,
and Shuvalov, V. A., eds) [in Russian], Institute of Computer-Aided
Studies, Izhevsk, Vol. 1, pp. 407-460.
3.Mubarakshina, M. M., and Ivanov, B. N. (2010) The
production and scavenging of reactive oxygen species in the
plastoquinone pool of chloroplast thylakoid membranes, Physiol.
Plant., 140, 103-110, doi:
10.1111/j.1399-3054.2010.01391.x.
4.Pospíšil, P. (2012) Molecular
mechanisms of production and scavenging of reactive oxygen species by
photosystem II, Biochim. Biophys. Acta, 1817, 218-231,
doi: 10.1016/j.bbabio.2011.05.017.
5.Kozuleva, M. A., and Ivanov, B. N. (2016) The
mechanisms of oxygen reduction in the terminal reducing segment of the
chloroplast photosynthetic electron transport chain, Plant Cell
Physiol., 57, 1397-1404, doi: 10.1093/pcp/pcw035.
6.Kozuleva, M. A., Ivanov, B. N., Vetoshkina, D. V.,
and Borisova-Mubarakshina, M. M. (2020) Minimizing an electron flow to
molecular oxygen in photosynthetic electron transfer chain: an
evolutionary view, Front. Plant Sci., 11, 211, doi:
10.3389/fpls.2020.00211.
7.Sarewicz, M., Pintscher, S., Pietras, R., Borek,
A., Bujnowicz, Ł., Hanke, G., Cramer, W. A., Finazzi, G., and
Osyczka, A. (2021) Catalytic reactions and energy conservation in the
cytochrome bc1 and b6f complexes of
energy-transducing membranes, Chem. Rev., 121, 2020-2108,
doi: 10.1021/acs.chemrev.0c00712.
8.Allen, J. F., and Hall, D. O. (1973) Superoxide
reduction as a mechanism of ascorbate-stimulated oxygen uptake by
isolated chloroplasts, Biochem. Biophys. Res. Commun.,
52, 856-862, doi: 10.1016/0006-291X(73)91016-4.
9.Asada, K., Kiso, K., and Yoshikawa, K. (1974)
Univalent reduction of molecular oxygen by spinach chloroplasts on
illumination, J. Biol. Chem., 249, 2175-2181, doi:
10.1016/S0021-9258(19)42815-9.
10.Wardman, P. (1990) Bioreductive activation of
quinones: redox properties and thiol reactivity, Free Radic. Res.
Commun., 8, 219-229, doi: 10.3109/10715769009053355.
11.Takahashi, M., and Asada, K. (1988) Superoxide
production in aprotic interior of chloroplast thylakoids, Arch.
Biochem. Biophys., 267, 714-722, doi:
10.1016/0003-9861(88)90080-X.
12.Kozuleva, M., Klenina, I., Proskuryakov, I.,
Kirilyuk, I., and Ivanov, B. (2011) Production of superoxide in
chloroplast thylakoid membranes: ESR study with cyclic hydroxylamines
of different lipophilicity, FEBS Lett., 585, 1067-1071,
doi: 10.1016/j.febslet.2011.03.004.
13.Kozuleva, M., Klenina, I., Mysin, I., Kirilyuk,
I., Opanasenko, V., Proskuryakov, I., and Ivanov, B. (2015)
Quantification of superoxide radical production in thylakoid membrane
using cyclic hydroxylamines, Free Radic. Biol. Med., 89,
1014-1023, doi: 10.1016/j.freeradbiomed.2015.08.016.
14.Kozuleva, M., Goss, T., Twachtmann, M., Rudi, K.,
Trapka, J., Selinski, J., Ivanov, B., Garapati, P., Steinhoff, H. J.,
Hase, T., Scheibe, R., Klare, J. P., and Hanke, G. T. (2016)
Ferredoxin:NADP(H) oxidoreductase abundance and location influences
redox poise and stress tolerance, Plant Physiol., 172,
1480-1493, doi: 10.1104/pp.16.01084.
15.Fantuzzi, A., Allgöwer, F., Baker, H.,
McGuire, G., Teh, W. K., Gamiz-Hernandez, A. P., Kaila, V. R. I., and
Rutherford, A. W. (2022) Bicarbonate-controlled reduction of oxygen by
the QA semiquinone in Photosystem II in membranes, Proc. Natl. Acad.
Sci. USA, 119, e2116063119, doi:
10.1073/pnas.2116063119.
16.Khorobrykh, S. A., and Ivanov, B. N. (2002)
Oxygen reduction in a plastoquinone pool of isolated pea thylakoids,
Photosynth. Res., 71, 209-219, doi:
10.1023/A:1015583502345.
17.Ford, R. C., and Evans, M. C. W. (1983) Isolation
of a photosystem 2 preparation from higher plants with highly enriched
oxygen evolution activity, FEBS Lett., 160, 159-164, doi:
10.1016/0014-5793(83)80957-0.
18.Fan, D.-Y., Hope, A. B., Smith, P. J., Jia, H.,
Pace, R. J., Anderson, J. M., and Chow, W. S. (2007) The stoichiometry
of the two photosystems in higher plants revisited, Biochim.
Biophys. Acta Bioenerg., 1767, 1064-1072, doi:
10.1016/j.bbabio.2007.06.001.
19.Baniulis, D., Hasan, S. S., Stofleth, J. T., and
Cramer, W. A. (2013) Mechanism of enhanced superoxide production in the
cytochrome b6f complex of oxygenic
photosynthesis, Biochemistry, 52, 8975-8983, doi:
10.1021/bi4013534.
20.Kozuleva, M., Petrova, A., Milrad, Y., Semenov,
A., Ivanov, B., Redding, K. E., and Iftach, Y. (2021) Phylloquinone is
the principal Mehler reaction site within photosystem I in high light,
Plant Physiol., 186, 1848-1858, doi:
10.1093/plphys/kiab221.
21.Hosein, B., and Palmer, G. (1983) The kinetics
and mechanism of oxidation of reduced spinach ferredoxin by molecular
oxygen and its reduced products, Biochim. Biophys. Acta
Bioenerg., 723, 383-390, doi:
10.1016/0005-2728(83)90045-2.
22.Golbeck, J., and Radmer, R. (1984) Is the rate of
oxygen uptake by reduced ferredoxin sufficient to account for
photosystem I-mediated O2 reduction, Adv. Photosynth.
Res., 1, 561.
23.Böhme, H. (1978) Quantitative determination
of ferredoxin, ferredoxin-NADP+ reductase and plastocyanin
in spinach chloroplasts, Eur. J. Biochem., 83, 137-141,
doi: 10.1111/j.1432-1033.1978.tb12077.x.
24.McKenzie, S. D., Ibrahim, I. M., Aryal, U. K.,
and Puthiyaveetil, S. (2020) Stoichiometry of protein complexes in
plant photosynthetic membranes, Biochim. Biophys. Acta
Bioenerg., 1861, 148141, doi:
10.1016/j.bbabio.2019.148141.
25.Frankel, L. K., Sallans, L., Limbach, P. A., and
Bricker, T. M. (2013) Oxidized amino acid residues in the vicinity of
QA and PheoD1 of the photosystem II reaction center: putative
generation sites of reducing-side reactive oxygen species, PLoS
One, 8, e58042, doi: 10.1371/journal.pone.0058042.
26.Kale, R., Hebert, A. E., Frankel, L. K., Sallans,
L., Bricker, T. M., and Pospíšil, P. (2017) Amino acid
oxidation of the D1 and D2 proteins by oxygen radicals during
photoinhibition of Photosystem II, Proc. Natl. Acad. Sci. USA,
114, 2988-2993, doi: 10.1073/pnas.1618922114.
27.Kumar, A., Prasad, A.,
Sedlářová, M., Kale, R., Frankel, L. K., Sallans,
L., Bricker, T. M., and Pospíšil, P. (2021) Tocopherol
controls D1 amino acid oxidation by oxygen radicals in Photosystem II,
Proc. Natl. Acad. Sci. USA, 118, e2019246118, doi:
10.1073/pnas.2019246118.
28.Taylor, R. M., Sallans, L., Frankel, L. K., and
Bricker, T. M. (2018) Natively oxidized amino acid residues in the
spinach cytochrome b6f complex, Photosynth.
Res., 137, 141-151, doi: 10.1007/s11120-018-0485-0.
29.Ananyev, G., Renger, G., Wacker, U., and Klimov,
V. (1994) The photoproduction of superoxide radicals and the superoxide
dismutase activity of Photosystem II. The possible involvement of
cytochrome b559, Photosynth. Res., 41, 327-338,
doi: 10.1007/BF00019410.
30.Cleland, R. E., and Grace, S. C. (1999)
Voltammetric detection of superoxide production by photosystem II,
FEBS Lett., 457, 348-352, doi:
10.1016/S0014-5793(99)01067-4.
31.Brinkert, K., Causmaecker, S. D.,
Krieger-Liszkay, A., Fantuzzi, A., and Rutherford, A. W. (2016)
Bicarbonate-induced redox tuning in Photosystem II for regulation and
protection, Proc. Natl. Acad. Sci. USA, 113, 12144-12149,
doi: 10.1073/pnas.1608862113.
32.Linke, K., and Ho, F. M. (2014) Water in
photosystem II: structural, functional and mechanistic considerations,
Biochim. Biophys. Acta Bioenerg., 1837, 14-32, doi:
10.1016/j.bbabio.2013.08.003.
33.Causmaecker, S. D., Douglass, J. S., Fantuzzi,
A., Nitschke, W., and Rutherford, A. W. (2019) Energetics of the
exchangeable quinone, QB, in Photosystem II, Proc. Natl.
Acad. Sci. USA, 116, 19458-19463, doi:
10.1073/pnas.1910675116.
34.Kruk, J., and Strzałka, K. (1999) Dark
reoxidation of the plastoquinone-pool is mediated by the low-potential
form of cytochrome b-559 in spinach thylakoids, Photosynth.
Res., 62, 273-279, doi: 10.1023/A:1006374319191.
35.Pospíšil, P.,
Šnyrychová, I., Kruk, J., Strzałka, K., and
Nauš, J. (2006) Evidence that cytochrome b559 is involved
in superoxide production in photosystem II: effect of synthetic
short-chain plastoquinones in a cytochrome b559 tobacco mutant,
Biochem. J., 397, 321-327, doi: 10.1042/BJ20060068.
36.Müh, F., and Zouni, A. (2016) Cytochrome
b 559 in Photosystem II, in Adv. Photosynth.
Respir. Springer, Dordrecht, 41, 143-175, doi:
10.1007/978-94-017-7481-9_8.
37.Shuvalov, V. A., Schreiber, U., and Heber, U.
(1994) Spectral and thermodynamic properties of the two hemes of the
D1D2cytochrome b-559 complex of spinach, FEBS Lett.,
337, 226-230, doi: 10.1016/0014-5793(94)80196-7.
38.Yadav, D. K., Prasad, A., Kruk, J., and
Pospíšil, P. (2014) Evidence for the involvement of
loosely bound plastosemiquinones in superoxide anion radical production
in photosystem II, PLoS One, 9, e115466, doi:
10.1371/journal.pone.0115466.
39.Khorobrykh, A. (2019) Hydrogen peroxide and
superoxide anion radical photoproduction in PSII preparations at
various modifications of the water-oxidizing complex, Plants,
8, 329, doi: 10.3390/plants8090329.
40.Mubarakshina, M., Khorobrykh, S., and Ivanov, B.
(2006) Oxygen reduction in chloroplast thylakoids results in production
of hydrogen peroxide inside the membrane, Biochim. Biophys. Acta
Bioenerg., 1757, 1496-1503, doi:
10.1016/j.bbabio.2006.09.004.
41.McCauley, S. W., and Melis, A. (1986)
Quantitation of plastoquinone photoreduction in spinach chloroplasts,
Photosynth. Res., 8, 3-16, doi: 10.1007/BF00028472.
42.Khorobrykh, S., Mubarakshina, M., and Ivanov, B.
(2004) Photosystem I is not solely responsible for oxygen reduction in
isolated thylakoids, Biochim. Biophys. Acta Bioenerg.,
1657, 164-167, doi: 10.1016/j.bbabio.2004.04.009.
43.Forquer, I., Covian, R., Bowman, M. K.,
Trumpower, B. L., and Kramer, D. M. (2006) Similar transition states
mediate the Q-cycle and superoxide production by the cytochrome
bc1 complex, J. Biol. Chem., 281,
38459-38465, doi: 10.1074/jbc.M605119200.
44.Vetoshkina, D. V., Ivanov, B. N., Khorobrykh, S.
A., Proskuryakov, I. I., and Borisova-Mubarakshina, M. M. (2017)
Involvement of the chloroplast plastoquinone pool in the Mehler
reaction, Physiol. Plant., 161, 45-55, doi:
10.1111/ppl.12560.
45.Tikhonov, A. N. (2014) The cytochrome
b6f complex at the crossroad of photosynthetic
electron transport pathways, Plant Physiol. Biochem., 81,
163-183, doi: 10.1016/j.plaphy.2013.12.011.
46.Kramer, D. M., Crofts, A. R. (1994)
Re-examination of the properties and function of the b cytochromes of
the thylakoid cytochrome bf complex, Biochim. Biophys. Acta
Bioenerg., 1184, 193-201, doi:
10.1016/0005-2728(94)90223-2.
47.Sang, M., Qin, X. C., Wang, W. D., Xie, J., Chen,
X. B., Wang, K. B., Zhang, J. P., Li, L. B., Kuang, T. Y. (2011)
High-light-induced superoxide anion radical formation in cytochrome
b6f complex from spinach as detected by EPR
spectroscopy, Photosynthetica, 49, 48-54, doi:
10.1007/s11099-011-0008-0.
48.Šnyrychová, I.,
Pospíšil, P., and Nauš, J. (2006) Reaction
pathways involved in the production of hydroxyl radicals in thylakoid
membrane: EPR spin-trapping study, Photochem. Photobiol. Sci.,
5, 472-476, doi: 10.1039/B514394B.
49.Kozuleva, M. A., Naidov, I. A., Mubarakshina, M.
M., and Ivanov, B. N. (2007) Participation of ferredoxin in oxygen
reduction by the photosynthetic electron transport chain,
Biophysics, 52, 393-397, doi:
10.1134/S0006350907040069.
50.Badger, M. R. (1985) Photosynthetic oxygen
exchange, Annu. Rev. Plant. Physiol., 36, 27-53, doi:
10.1146/annurev.pp.36.060185.000331.
51.Allen, J. F. (1975) Oxygen reduction and optimum
production of ATP in photosynthesis, Nature, 256,
599-600, doi: 10.1038/256599a0.
52.Furbank, R. T., and Badger, M. R. (1983) Oxygen
exchange associated with electron transport and photophosphorylation in
spinach thylakoids, Biochim. Biophys. Acta Bioenerg.,
723, 400-409, doi: 10.1016/0005-2728(83)90047-6.
53.Kozuleva, M. A., and Ivanov, B. N. (2010)
Evaluation of the participation of ferredoxin in oxygen reduction in
the photosynthetic electron transport chain of isolated pea thylakoids,
Photosynth. Res., 105, 51-61, doi:
10.1007/s11120-010-9565-5.
54.Asada, K. (1999) The water–water cycle in
chloroplasts: scavenging of active oxygens and dissipation of excess
photons, Annu. Rev. Plant. Physiol. Plant. Mol. Biol.,
50, 601-639, doi: 10.1146/annurev.arplant.50.1.601.
55.Asada, K., and Nakano, Y. (1978) Affinity for
oxygen in photoreduction of molecular oxygen and scavenging of hydrogen
peroxide in spinach chloroplasts, Photochem. Photobiol.,
28, 917-920, doi: 10.1111/j.1751-1097.1978.tb07040.x.
56.Petrova, A., Mamedov, M., Ivanov, B., Semenov,
A., and Kozuleva, M. (2018) Effect of artificial redox mediators on the
photoinduced oxygen reduction by photosystem I complexes,
Photosynth. Res., 137, 421-429, doi:
10.1007/s11120-018-0514-z.
57.Robinson, J. M. (1988) Does O2
photoreduction occur within chloroplasts in vivo? Physiol.
Plant., 72, 666-680, doi:
10.1111/j.1399-3054.1988.tb09181.x.
58.Miyake, C., Schreiber, U., Hormann, H., Sano, S.,
and Asada, K. (1998) The FAD-enzyme monodehydroascorbate radical
reductase mediates photoproduction of superoxide radicals in spinach
thylakoid membranes, Plant Cell Physiol., 39, 821-829,
doi: 10.1093/oxfordjournals.pcp.a029440.
59.Hanke, G. T., Endo, T., Satoh, F., and Hase, T.
(2008) Altered photosynthetic electron channelling into cyclic electron
flow and nitrite assimilation in a mutant of ferredoxin:NADP(H)
reductase, Plant Cell Environ., 31, 1017-1028, doi:
10.1111/j.1365-3040.2008.01814.x.
60.Kramer, M., Rodriguez-Heredia, M., Saccon, F.,
Mosebach, L., Twachtmann, M., Krieger-Liszkay, A., Duffy, C., Knell, R.
J., Finazzi, G., and Hanke, G. T. (2021) Regulation of photosynthetic
electron flow on dark to light transition by ferredoxin:NADP(H)
oxidoreductase interactions, ELife, 10, e56088, doi:
10.7554/eLife.56088.
61.Buchert, F., Mosebach, L., Gäbelein, P., and
Hippler, M. (2020) PGR5 is required for efficient Q cycle in the
cytochrome b6f complex during cyclic electron flow,
Biochem. J., 477, 1631-1650, doi:
10.1042/BCJ20190914.
62.Malone, L. A., Proctor, M. S., Hitchcock, A.,
Hunter, C. N., and Johnson, M. P. (2021) Cytochrome
b6f – orchestrator of photosynthetic electron
transfer, Biochim. Biophys. Acta Bioenerg., 1862, 148380,
doi: 10.1016/j.bbabio.2021.148380.
63.Kozuleva, M. (2022) Recent advances in the
understanding of superoxide anion radical formation in the
photosynthetic electron transport chain, Acta Physiol. Plant.,
44, 92, doi: 10.1007/s11738-022-03428-0.
64.Hiyama, T., and Ke, B. (1971) A new
photosynthetic pigment, “P430”: its possible role as the
primary electron acceptor of photosystem I, Proc. Natl. Acad. Sci.
USA, 68, 1010-1013, doi: 10.1073/pnas.68.5.1010.
65.Kozuleva, M. A., Vetoshkina, D. V., Petrova, A.
A., Borisova-Mubarakshina, M. M., and Ivanov, B. N. (2015) The study of
oxygen reduction in photosystem I of higher plants using electron
donors for this photosystem in intact thylakoids, Biochemistry
(Moscow) Suppl. Ser. A Membr. Cell Biol., 9, 246-251, doi:
10.1134/S1990747814060026.
66.Khorobrykh, S., and Tyystjärvi, E. (2018)
Plastoquinol generates and scavenges reactive oxygen species in organic
solvent: potential relevance for thylakoids, Biochim. Biophys. Acta
Bioenerg., 1859, 1119-1131, doi:
10.1016/j.bbabio.2018.07.003.
67.Takahashi, M., and Asada, K. (1982) Dependence of
oxygen affinity for Mehler reaction on photochemical activity of
chloroplast thylakoids, Plant Cell Physiol., 23,
1457-1461, doi: 10.1093/oxfordjournals.pcp.a076495.
68.Kruk, J., Jemioła-Rzemińska, M.,
Burda, K., Schmid, G. H., and Strzałka, K. (2003) Scavenging of
superoxide generated in photosystem I by plastoquinol and other
prenyllipids in thylakoid membranes, Biochemistry, 42,
8501-8505, doi: 10.1021/bi034036q.
69.Kozuleva, M. A., Petrova, A. A., Mamedov, M. D.,
Semenov, A. Yu., and Ivanov, B. N. (2014) O2 reduction by
photosystem I involves phylloquinone under steady-state illumination,
FEBS Lett., 588, 4364-4368, doi:
10.1016/j.febslet.2014.10.003.
70.Semenov, A. Y., Vassiliev, I. R., van der Est,
A., Mamedov, M. D., Zybailov, B., Shen, G., Stehlik, D., Diner, B. A.,
Chitnis, P. R., and Golbeck, J. H. (2000) Recruitment of a foreign
quinone into the A1 site of Photosystem I: altered kinetics
of electron transfer in phylloquinone biosynthetic pathway mutants
studied by time-resolved optical, EPR, and electrometric techniques,
J. Biol. Chem., 275, 23429-23438, doi:
10.1074/jbc.M000508200.
71.Santabarbara, S., Bullock, B., Rappaport, F., and
Redding, K. E. (2015) Controlling electron transfer between the two
cofactor chains of photosystem I by the redox state of one of their
components, Biophys. J., 108, 1537-1547, doi:
10.1016/j.bpj.2015.01.009.
72.Kale, R., Sallans, L., Frankel, L. K., and
Bricker, T. M. (2020) Natively oxidized amino acid residues in the
spinach PS I-LHC I supercomplex, Photosynth. Res., 143,
263-273, doi: 10.1007/s11120-019-00698-7.
73.Milanovsky, G. E., Petrova, A. A., Cherepanov, D.
A., and Semenov, A. Yu. (2017) Kinetic modeling of electron transfer
reactions in photosystem I complexes of various structures with
substituted quinone acceptors, Photosynth. Res., 133,
185-199, doi: 10.1007/s11120-017-0366-y.
74.Ivanov, B. (2000) The competition between methyl
viologen and monodehydroascorbate radical as electron acceptors in
spinach thylakoids and intact chloroplasts, Free Radic. Res.,
33, 217-227, doi: 10.1080/10715760000301391.
75.Bukhov, N. G., Govindachary, S., Egorova, E. A.,
Joly, D., and Carpentier, R. (2003)
N,N,N′,N′-tetramethyl-p-phenylenediamine initiates the
appearance of a well-resolved I peak in the kinetics of chlorophyll
fluorescence rise in isolated thylakoids, Biochim. Biophys. Acta
Bioenerg., 1607, 91-96, doi:
10.1016/j.bbabio.2003.09.002.
76.Trubitsin, B. V., Mamedov, M. D., Semenov, A.
Yu., and Tikhonov, A. N. (2014) Interaction of ascorbate with
photosystem I, Photosynth. Res., 122, 215-231, doi:
10.1007/s11120-014-0023-7.
77.Michelet, L., and Krieger-Liszkay, A. (2012)
Reactive oxygen intermediates produced by photosynthetic electron
transport are enhanced in short-day grown plants, Biochim. Biophys.
Acta Bioenerg., 1817, 1306-1313, doi:
10.1016/j.bbabio.2011.11.014.
78.Krieger-Liszkay, A., Shimakawa, G., and
Sétif, P. (2020) Role of the two PsaE isoforms on O2
reduction at photosystem I in Arabidopsis thaliana, Biochim.
Biophys. Acta Bioenerg., 1861, 148089, doi:
10.1016/j.bbabio.2019.148089.
79.Marco, P., Elman, T., and Yacoby, I. (2019)
Binding of ferredoxin NADP+ oxidoreductase (FNR) to plant
photosystem I, Biochim. Biophys. Acta Bioenerg., 1860,
689-698, doi: 10.1016/j.bbabio.2019.07.007.
80.Andersen, B., Scheller, H. V., and Møller,
B. L. (1992) The PSI-E subunit of photosystem I binds
ferredoxin:NADP+ oxidoreductase, FEBS Lett.,
311, 169-173, doi: 10.1016/0014-5793(92)81391-X.
81.Benz, J. P., Stengel, A., Lintala, M., Lee,
Y.-H., Weber, A., Philippar, K., Gügel, I. L., Kaieda, S.,
Ikegami, T., Mulo, P., Soll, J., and Bölter, B. (2009)
Arabidopsis Tic62 and ferredoxin-NADP(H) oxidoreductase form
light-regulated complexes that are integrated into the chloroplast
redox poise, Plant Cell, 21, 3965-3983, doi:
10.1105/tpc.109.069815.
82.Jurić, S., Hazler-Pilepić, K.,
Tomašić, A., Lepeduš, H., Jeličić,
B., Puthiyaveetil, S., Bionda, T., Vojta, L., Allen, J. F., Schleiff,
E., and Fulgosi, H. (2009) Tethering of ferredoxin:NADP+
oxidoreductase to thylakoid membranes is mediated by novel chloroplast
protein TROL, Plant J., 60, 783-794, doi:
10.1111/j.1365-313X.2009.03999.x.
83.Jagannathan, B., Shen, G., and Golbeck, J. H.
(2012) The evolution of type I reaction centers: the response to
oxygenic photosynthesis, in Functional Genomics and Evolution of
Photosynthetic Systems, Springer, Dordrecht, pp. 285-316, doi:
10.1007/978-94-007-1533-2_12.
84.Rutherford, A. W., Osyczka, A., and Rappaport, F.
(2012) Back-reactions, short-circuits, leaks and other energy wasteful
reactions in biological electron transfer: Redox tuning to survive life
in O2, FEBS Lett., 586, 603-616, doi:
10.1016/j.febslet.2011.12.039.
85.Pierella Karlusich, J. J., and Carrillo, N.
(2017) Evolution of the acceptor side of photosystem I: ferredoxin,
flavodoxin, and ferredoxin-NADP+ oxidoreductase,
Photosyn. Res., 134, 235-250, doi:
10.1007/s11120-017-0338-2.
86.Orf, G. S., Gisriel, C., and Redding, K. E.
(2018) Evolution of photosynthetic reaction centers: insights from the
structure of the heliobacterial reaction center, Photosynth.
Res., 138, 11-37, doi: 10.1007/s11120-018-0503-2.
87.Hanke, G., and Mulo, P. (2013) Plant type
ferredoxins and ferredoxin-dependent metabolism, Plant Cell
Environ., 36, 1071-1084, doi: 10.1111/pce.12046.
88.Fischer, N., Sétif, P., and Rochaix, J.-D.
(1997) Targeted mutations in the psaC gene of Chlamydomonas
reinhardtii: preferential reduction of FB at low temperature is not
accompanied by altered electron flow from photosystem I to ferredoxin,
Biochemistry, 36, 93-102, doi: 10.1021/bi962244v.
89.Shinkarev, V. P., Vassiliev, I. R., and Golbeck,
J. H. (2000) A kinetic assessment of the sequence of electron transfer
from F(X) to F(A) and further to F(B) in photosystem I: the value of
the equilibrium constant between F(X) and F(A), Biophys. J.,
78, 363-372, doi: 10.1016/S0006-3495(00)76599-4.
90.Ptushenko, V. V., Cherepanov, D. A., Krishtalik,
L. I., and Semenov, A. Y. (2008) Semi-continuum electrostatic
calculations of redox potentials in photosystem I, Photosynth.
Res., 97, 55-74, doi: 10.1007/s11120-008-9309-y.
91.Schoepp-Cothenet, B., van Lis, R., Atteia, A.,
Baymann, F., Capowiez, L., Ducluzeau, A.-L., Duval, S., Brink, F.,
Russell, M. J., and Nitschke, W. (2013) On the universal core of
bioenergetics, Biochim. Biophys. Acta Bioenerg., 1827,
79-93, doi: 10.1016/j.bbabio.2012.09.005.
92.Massey, V. (1994) Activation of molecular oxygen
by flavins and flavoproteins, J. Biol. Chem., 269,
22459-22462, doi: 10.1016/S0021-9258(17)31664-2.
93.Ceccarelli, E. A., Arakaki, A. K., Cortez, N.,
and Carrillo, N. (2004) Functional plasticity and catalytic efficiency
in plant and bacterial ferredoxin-NADP(H) reductases, Biochim.
Biophys. Acta Proteins Proteomics, 1698, 155-165, doi:
10.1016/j.bbapap.2003.12.005.
94.Carrillo, N., and Ceccarelli, E. A. (2003) Open
questions in ferredoxin-NADP+ reductase catalytic mechanism,
Eur. J. Biochem., 270, 1900-1915, doi:
10.1046/j.1432-1033.2003.03566.x.
95.Batie, C. J., and Kamin, H. (1984)
Ferredoxin:NADP+ oxidoreductase. Equilibria in binary and
ternary complexes with NADP+ and ferredoxin, J. Biol.
Chem., 259, 8832-8839, doi:
10.1016/S0021-9258(17)47229-2.
96.Mulo, P., and Medina, M. (2017) Interaction and
electron transfer between ferredoxin-NADP+ oxidoreductase
and its partners: structural, functional, and physiological
implications, Photosynth. Res., 134, 265-280, doi:
10.1007/s11120-017-0372-0.
97.Drachev, L. A., Kaurov, B. S., Mamedov, M. D.,
Mulkidjanian, A. Y., Semenov, A. Yu, Shinkarev, V. P., Skulachev, V.
P., and Verkgovsky, M. I. (1989) Flash-induced electrogenic events in
the photosynthetic reaction center and bc1 complexes
of Rhodobacter sphaeroides chromatophores, Biochim. Biophys.
Acta, 973, 189-197, doi: 10.1016/S0005-2728(89)80421-9.
98.De Vries, S., Berden, J. A., and Slater, E. C.
(1980) Properties of a semiquinone anion located in the
QH2:cytochrome c oxidoreductase segment of the
mitochondrial respiratory chain, FEBS Lett., 122,
143-148, doi: 10.1016/0014-5793(80)80422-4.
99.Stroebel, D., Choquet, Y., Popot, J.-L., and
Picot, D. (2003) An atypical haem in the cytochrome b(6)f
complex, Nature, 426, 413-418, doi:
10.1038/nature02155.
100.Vilyanen, D., Naydov, I., Ivanov, B.,
Borisova-Mubarakshina, M., and Kozuleva, M. (2022) Inhibition of
plastoquinol oxidation at the cytochrome b6f complex
by dinitrophenyl ether of iodonitrothymol (DNP-INT) depends on
irradiance and H+ uptake by thylakoid membranes, Biochim.
Biophys. Acta Bioenerg., 1863, 148506, doi:
10.1016/j.bbabio.2021.148506.
101.Schoepp-Cothenet, B., Lieutaud, C., Baymann,
F., Verméglio, A., Friedrich, T., Kramer, D. M., and Nitschke,
W. (2009) Menaquinone as pool quinone in a purple bacterium, Proc.
Natl. Acad. Sci. USA, 106, 8549-8554, doi:
10.1073/pnas.0813173106.
102.Bergdoll, L., ten Brink, F., Nitschke, W.,
Picot, D., and Baymann, F. (2016) From low- to high-potential
bioenergetic chains: thermodynamic constraints of Q-cycle function,
Biochim. Biophys. Acta Bioenerg., 1857, 1569-1579, doi:
10.1016/j.bbabio.2016.06.006.
103.Alric, J., Pierre, Y., Picot, D., Lavergne, J.,
and Rappaport, F. (2005) Spectral and redox characterization of the
heme ci of the cytochrome bf complex, Proc. Natl. Acad. Sci.
USA, 102, 15860-15865, doi: 10.1073/pnas.0508102102.
104.Gisriel, C., Sarrou, I., Ferlez, B., Golbeck,
J. H., Redding, K. E., and Fromme, R. (2017) Structure of a symmetric
photosynthetic reaction center-photosystem, Science, 357,
1021-1025, doi: 10.1126/science.aan5611.
105.He, Z., Ferlez, B., Kurashov, V., Tank, M.,
Golbeck, J. H., and Bryant, D. A. (2019) Reaction centers of the
thermophilic microaerophile, Chloracidobacterium
thermophilum (Acidobacteria) I: biochemical and biophysical
characterization, Photosynth. Res., 142, 87-103, doi:
10.1007/s11120-019-00650-9.
106.Su, X., Ma, J., Pan, X., Zhao, X., Chang, W.,
Liu, Z., Zhang, X., and Li, M. (2019) Antenna arrangement and energy
transfer pathways of a green algal photosystem-I-LHCI supercomplex,
Nat. Plants, 5, 273-281, doi:
10.1038/s41477-019-0380-5.
107.Kashey, T. S., Luu, D. D., Cowgill, J. C.,
Baker, P. L., and Redding, K. E. (2018) Light-driven quinone reduction
in heliobacterial membranes, Photosynth. Res., 138, 1-9,
doi: 10.1007/s11120-018-0496-x.
108.McConnell, M. D., Cowgill, J. B., Baker, P. L.,
Rappaport, F., and Redding, K. E. (2011) Double reduction of
plastoquinone to plastoquinol in photosystem 1, Biochemistry,
50, 11034-11046, doi: 10.1021/bi201131r.
109.Guergova-Kuras, M., Boudreaux, B., Joliot, A.,
Joliot, P., and Redding, K. (2001) Evidence for two active branches for
electron transfer in photosystem I, Proc. Natl. Acad. Sci. USA,
98, 4437-4442, doi: 10.1073/pnas.081078898.
110.Ksas, B., Alric, J., Caffarri, S., and Havaux,
M. (2022) Plastoquinone homeostasis in plant acclimation to light
intensity, Photosynth. Res., 152, 43-54, doi:
10.1007/s11120-021-00889-1.
111.Suslichenko, I. S., and Tikhonov, A. N. (2019)
Photo-reducible plastoquinone pools in chloroplasts of
Tradescentia plants acclimated to high and low light, FEBS
Lett., 593, 788-798, doi: 10.1002/1873-3468.13366.