Specific Cleavage of Hybrid Proteins by Proteinase Encoded by the KEX2 Gene
L. Ya. Bessmertnaya,1,2 I. I. Loiko,1 T. I. Goncharova,1 N. V. Ivanov,1 L. D. Rumsh,1 and V. K. Antonov1
1Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117871 Russia; fax: (095) 335-7103; E-mail: mila@enzyme.siobc.ras.ru2To whom correspondence should be addressed.
Submitted February 6, 1997; revision submitted March 17, 1997.
A method for isolation of the KEX2-gene-encoded membrane-bound proteinase from alpha-cells of Saccharomyces cerevisiae yeast has been modified. The isolated enzyme hydrolyzes peptides and proteins with basic amino acid pairs which are cleaved at the C-ends of their peptide bonds. Because KEX2 proteinase is located within the Golgi compartment, it may be isolated by differential centrifugation of broken cells at 7000g for 15 min and at 20,000g for 15 min. By extracting the fraction that contains the active enzyme by a detergent solution, a protein has been obtained with specific activity 30 times higher than that of the membrane extract prepared according to the standard technique. This protocol decreases the number of steps required to isolate the enzyme. The effects of pH and inhibitors on KEX2 proteinase-catalyzed hydrolysis of Ac-Leu-Lys-Arg-pNA were studied. KEX2 proteinase can participate in peptide hormone processing because it cleaves human proinsulin at the peptide bond between Arg32 and Glu33. The KEX2 proteinase can specifically cleave large recombinant proteins, for example, a protein consisting of a gamma-interferon fragment linked to HIV1-proteinase via a Lys-Arg-containing peptide.
KEY WORDS: KEX2 proteinase, protein processing, proinsulin, recombinant proteins, proteolysis.
Abbreviations: DFP) diisopropyl fluorophosphate; PMSF) phenylmethylsulfonyl fluoride; Ac-Leu-Lys-Arg-pNA) acetyl-L-leucyl-L-lysyl-L-arginine-para-nitroanilide; Ac-Leu-Leu-Arg-pNA) acetyl-L-leucyl-L-leucyl-para-nitroanilide; TFA) trifluoroacetic acid; BAPNA) benzoyl-L-arginine-para-nitroanilide; ATEE) acetyl-L-tyrosine ethyl ether; Hip-Arg) hippuryl-L-arginine; LTI) bovine lung trypsin inhibitor; STI) soybean trypsin inhibitor; UTI) human urine trypsin inhibitor.
One of the tasks of modern biochemistry is to elucidate the mechanisms
of action of enzymes known as convertases; they directly participate in
transforming the precursors of biologically active agents into their
functional forms. KEX2 proteinase of Saccharomyces cerevisiae
cells is a typical enzyme of such processing and may serve as a
simple model to study the conversion of prohormones in higher
eukaryotes [1-4]. The enzyme is
active toward pairs of basic amino acids within polypeptide chains and
cleaves peptide bonds at the C-end of these pairs. In peptide
substrates, KEX2 proteinase hydrolyzes peptide bonds downstream from
-Lys-Arg- and -Arg-Arg- pairs with similar efficacy and does not
hydrolyze -Lys-Lys- bonds [1-6]. It has been shown [7-10] that the hydrolysis of synthetic
high-molecular-weight substrates by yeast KEX2 proteinase and similar
proteinases depends upon the composition of the amino acid sequences
around the site of hydrolysis. For example, the presence of Arg in the
P4 position relative to the bond to be cleaved in a synthetic peptide
substrates accelerates its hydrolysis by some KEX2-like mammal
endoproteinases hundreds of times while inhibiting its hydrolysis by
yeast KEX2 proteinase [7, 10].
Studies of the KEX2 proteinase-mediated cleavage of artificial protein
constructs comprising beta-endorphin, calcitonin, and epidermal
growth factor with the upstream leader alpha-factor sequence
have shown that the absence of -Glu-Ala-Glu-Ala- tetrapeptide
downstream from the cleaved bond affects the rate of hydrolysis [11-13].
The objective of this work was to elucidate the capacity of yeast KEX2 proteinase in cleaving peptide chains that contain a pair of positively charged amino acids using proinsulin and recombinant proteins as models and to determine the optimal conditions for these reactions, i.e., the duration of hydrolysis, the concentrations of components, and the ratio of the enzyme and its substrate. The content of KEX2 proteinase in yeast cells is known to be less than 1·10-5 of total cell protein [2] making the isolation of this enzyme a difficult task. So, another purpose of this work was to improve the method generally used to isolate and to purify the enzyme.
MATERIALS AND METHODS
The following reagents were used: Tris and acetonitrile from Merck (USA); acrylamide, N,N'-methylene-bis-acrylamide, ammonium persulfate, and Protein Assay reagent from Bio-Rad (USA); glycine, urea, beta-mercaptoethanol, SDS, Coomassie G-250, and EDTA from Serva (Germany); protein standards for PAGE from Pharmacia (Sweden); Immobilon from Millipore (USA); Lubrol and LTI3 from Sigma (USA); DFP from Fluka (Switzerland); PMSF, STI, and ATEE from Reanal (Hungary). Other reagents were Russian products of the highest grade available.
Haploid Saccharomyces cerevisiae cells (X-2180 strain) were grown under anaerobic conditions at 30°C in 30 liters of media containing 1% yeast extract, 2% peptone, and 2% glucose. All subsequent procedures were performed at 4°C. The cells grown to 320 g were sedimented by centrifugation at 10,000g for 20 min, washed with 0.9% NaCl, and resuspended in 0.1 M sodium phosphate buffer, pH 7.0. The cells were frozen at -70°C, thawed, and broken by passing though a Gaulin flow disintegrator (USA).
To optimize the conditions of yeast cell disintegration with lysamidase (manufactured in Vyshnii Volochek), the reaction was performed with different enzyme concentrations in 25% sucrose or 1 M NaCl with subsequent dilutions of the solutions. Samples were withdrawn periodically, and cell lysis was monitored by spectrophotometry at 540 nm. Also, the percent of lysed cells was determined by microscopy. At yeast cell content corresponding to 100 mg/ml, the optimum duration of cell disintegration in the presence of 2 mg/ml lysamidase was 3 h.
Unbroken cells were separated by centrifugation at 5000g for 15 min, and the supernatant was further centrifuged at 7000g for 15 min. The final supernatant was centrifuged at 20,000g for 15 min. The sediment so obtained was washed four times with Tris-HCl buffer (10 mM, pH 7.0) and with the same buffer containing 0.5 M NaCl, each wash followed by centrifugation at 100,000g for 10 min. The washed sediment was extracted with a minimal volume of 10 mM Tris-HCl buffer (pH 7.0) containing 0.1 M NaCl and 1% Lubrol. The extract was diluted fivefold with 10 mM Tris-HCl buffer (pH 8.0), applied to a 5.0 × 1.0 cm DEAE-Servacel-filled column, and eluted with the same buffer with a linear gradient of NaCl (0.05-0.45 M).
During the purification KEX2 enzymatic activity was tested using a synthetic substrate, Ac-Leu-Lys-Arg-pNA. The accompanying trypsin- and chymotrypsin-like and carboxypeptidase activities were checked with BAPNA, ATEE, Hip-Arg, and Ac-Leu-Leu-Arg-pNA as substrates. Ac-Leu-Lys-Arg-pNa and Ac-Leu-Leu-Arg-pNA were synthesized in the Laboratory of Proteolytic Enzyme Chemistry.
Active fractions were dialyzed against 20 mM Tris-HCl buffer, pH 7.0, containing 0.01% Lubrol and 5 mM CaCl2 and concentrated using a Speedvac concentrator (USA). The concentrated fractions eluted from the ion-exchange column were applied to an affinity chromatography column (1.5 × 4 cm) filled with arginine-Sepharose and equilibrated with 20 mM Tris-HCl buffer, pH 7.0, containing 0.01% Lubrol. The column was washed with the same buffer, and the active enzyme was eluted with a 1 M NaCl-containing buffer or using a linear NaCl gradient, i.e., from 0.05 to 0.5 M. The fractions obtained were desalted on a PD2 column and concentrated.
A portion of the active fraction was applied to an affinity chromatography column (1.2 × 2.5 cm) filled with Sepharose 4B to which human urine trypsin inhibitor was bound (the inhibitor was a generous gift from Dr. O. G. Ogloblina of the Russian Cardiological Research Center, Moscow). The column was equilibrated with 20 mM Tris-HCl buffer, pH 7.0, containing 5 mM CaCl2. Elution was performed using 0.1 M sodium formate buffer, pH 4.0, containing 0.5 M NaCl.
Discontinuous glycerol density gradient was performed in six centrifuge tubes of 5 ml capacity by making consecutive 0.5 ml layers of 63% and 43% glycerol and 1 ml layers of 30% and 20% glycerol finally overlaid by 1 ml of sample in 10% glycerol and 1 ml of the buffer. All layers contained 20 mM Tris HCl (pH 7.0) and 5 mM CaCl2. After centrifugation in a L5-50 Beckman centrifuge (SW-50 rotor) at 45,000 rpm for 5 h, fractions were collected from the bottom using a peristaltic pump, and their optical densities at 280 nm and activities toward Ac-Leu-Lys-Arg-pNA and BAPNA were determined. Glycerol percent was measured by refractometry.
To determine enzymatic activity, an aliquot of enzyme preparation was transferred to 0.5 M Tris-HCl buffer, pH 7.0, with 10 mM CaCl2, after which 40 µl of either 10-2 M Ac-Leu-Lys-Arg-pNA or 10-2 M BAPNA was added to the solution. The final volume of this reaction mixture was 400 µl. The amount of p-nitroaniline formed in the reaction was determined with a Gilford spectrophotometer at 405 nm at 37°C. One unit of enzyme activity was taken as the amount of the enzyme that utilized 10 nmoles of its substrate per hour. Protein was determined according to Bradford [14].
Proinsulin hydrolysis by KEX2 proteinase was performed by adding an aliquot of the enzyme (0.2_2 units) to 60 µl of 0.2 M Tris-HCl buffer, pH 7.0, containing 2 mM CaCl2. The final volume was 100 µl. The final concentration of proinsulin in the reaction mixture varied from 0.2 to 1.8 mg/ml. The mixture was incubated at 37°C, and in 20, 40, 60, and 90 min after starting the reaction aliquots were taken and frozen.
The reaction mixture was analyzed (20-µl samples) by reverse-phase HPLC using 4.6 × 250 mm Lichrosorb C-8 columns (Beckman) and gradient elution with a mixture of 10% acetonitrile and 0.1% TFA in water (A) and 0.1% TFA in acetonitrile (B), the proportion of B in the mixture increasing from 15 to 40% over 20 min. The flow rate was 1 ml/min.
To determine the N-terminal amino acid generated from proinsulin by KEX2-catalyzed hydrolysis, the reaction mixture was analyzed after it was dansylated in 0.02 M ammonium acetate buffer, pH 7.1, containing 1 mM CaCl2.
A recombinant INF-link-HIV gene was constructed in the Laboratory of Proteolytic Enzyme Chemistry by A. Yu. Amerik and N. I. Dergousova. The corresponding hybrid protein was produced in E. coli where it was incorporated into inclusion bodies. The cells were broken by sonication, protein complexes were dissolved in 8 M urea, and products so obtained were dialyzed; then the recombinant protein was diluted to final content 1-2 mg/ml. The recombinant protein at concentrations in the range (4.5-9)·10-5 M was incubated at 37°C with KEX2 proteinase at concentrations in the range (1-2)·10-7 M in 20 mM Tris-HCl buffer, pH 7.0, containing 5 mM CaCl2. When the hybrid protein was hydrolyzed by the enzyme in the presence of 4 M urea, their concentrations were 4.5·10-4 and 2·10-6 M, respectively.
Gel electrophoresis was conducted according to Laemmli [15] in 1-mm thick 12% and 16% polyacrylamide gel plates with 4% concentrating gels. Electroblotting of protein bands obtained by electrophoresis was performed using a Bio-Rad apparatus (USA) at 11 V for 16 h in 0.025 M sodium bicarbonate (pH 9.0) with 10% methanol and 0.01% SDS. In this way proteins were transferred to Immobilon to determine their N-terminal amino acid sequences with the help of a 477A gas phase sequencer (Applied Biosystems).
RESULTS AND DISCUSSION
KEX2 proteinase was isolated according to the following scheme.
I. Yeast Cell Lysis. With a Gaulin disintegrator, this process requires multiple passages of the yeast cell suspension though the nozzle of the device, the output of broken cells never exceeding 50%. To increase the output, a stage of cell wall pre-exposure to lysamidase was introduced; this resulted in an appropriate degree of cell destruction reaching 90% (see "Materials and Methods").
II. Membrane Protein Centrifugation and Treatment with Lubrol. The centrifugation stage of KEX2 proteinase isolation procedure employed in this work differs from that described in the literature [1, 16, 17]. According to Mizuno et al. [1, 16], the disintegration of cells and separation of unbroken cells and cell walls (5000g, 20 min) is followed by centrifugation of the supernatant at 80,000g for 20 min. Membrane protein material obtained according to this protocol (method 1) by extracting the sediment with 1% Lubrol was found to be contaminated by other enzyme activities and to possess a low specific activity of the enzyme in question (0.38 U/mg protein), i.e., it was but a crude membrane preparation (Table 1).
TABLE 1. Purification of KEX2 Proteinase by
Method 1

Proceeding from the fact that KEX2 proteinase is localized mainly in the Golgi apparatus [17-19] and from regimes used to sediment intracellular components [20], we centrifuged broken cells in a series of experiments which revealed that the sediment obtained after centrifugation at 20,000g for 15 min of the supernatant prepared at 7000g for 15 min (method 2) contained the bulk of KEX2 proteinase with specific activity about 8-12 U/mg protein and virtually free of contaminating enzymes or only with trace amounts of them (Table 2). Lubrol extract of the sediment obtained by this differential centrifugation was used as a starting material to further purify the enzyme by chromatographic methods.
TABLE 2. Purification of KEX2 Proteinase by
Method 2

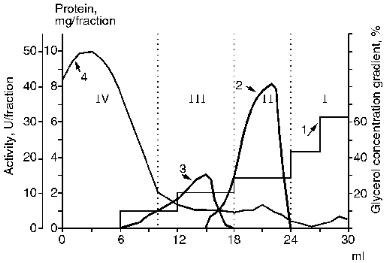
A still higher specific activity of membrane KEX2 proteinase extract was achieved after its high-speed centrifugation in a glycerol concentration gradient. In pilot experiments using a linear glycerol density gradient, it was found that the bulk of unwanted protein remained in the low density zone. By centrifuging samples of Lubrol extracts of membrane proteins at 200,000g for 5 h in a discontinuous gradient of glycerol concentration (from 10 to 63%), we were able to separate proteins with different phase states and different enzyme activities toward a variety of substrates (Fig. 1). The addition of a protein sample to a medial gradient zone causes the migration of the enzyme and solubilized contaminants in opposite directions, which precludes the nonspecific sorption of contaminants by the KEX2 enzyme preparation concentrating at the border between phases I and II. The discontinuous gradient centrifugation results in purified KEX2 proteinase preparation containing 70% of the initial enzymatic activity concentrates at the border of phases I and II and becomes separated from proteins with no activity that sediment to the bottom of the centrifuge tubes (fraction I) from a protein material active toward BAPNA (fraction III), and, also, from unwanted proteins that float to zone IV, which is characterized by a low glycerol concentration and consists mainly of the buffer solution. At this stage, the specific activity of the enzyme reaches 25 U/mg protein.
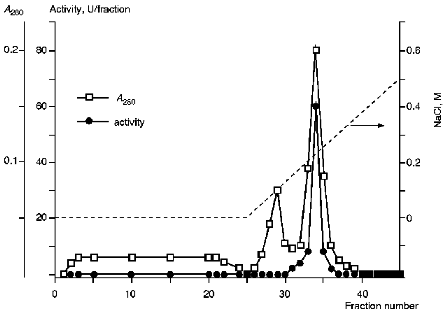
III. Chromatographic Purification of KEX2 Proteinase. Theenzyme was purified further by ion-exchange chromatography on DEAE-Servacel (Fig. 2). Active membrane fractions obtained by differential and density gradient centrifugation possessed specific activities that were 30 and, respectively, 60 times higher than that of the crude membrane extract (see Tables 1 and 2, stage 1). Therefore, the ion-exchange chromatography of these fractions (method 2, stage 2) yielded KEX2 protease preparations with specific activities that were also 30 times higher than the respective values of specific activities at the corresponding stage of method 1, i.e., stage 2.Fig. 1. KEX2 proteinase purification by centrifugation in a discontinuous glycerol density gradient. Fraction volume, 2 ml. 1) Initial discontinuous glycerol density gradient; 2) activity toward Ac-Leu-Lys-Arg-PNA; 3) activity toward BAPNA; 4) protein.
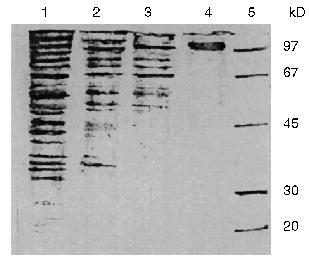
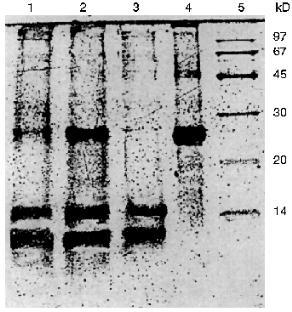
The additional purification of the proteinase by affinity chromatography on arginine-Sepharose (Fig. 3) or Sepharose with immobilized human urine trypsin inhibitor (UTI-Sepharose) [21-23] resulted in further increase in the specific activity of the enzyme, up to 260-300 U/mg protein, with virtually no loss of its total activity (Table 2). When KEX2 preparations so obtained were subjected to PAGE, they displayed one band corresponding to 100 kD (Fig. 4), in agreement with published data [2, 16].Fig. 2. DEAE-Servacel ion-exchange chromatography of membrane extract after its differential centrifugation by method 2. Fraction volume is 2 ml.
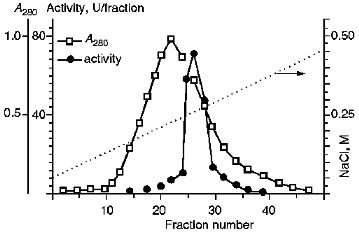
Fig. 3. KEX2 proteinase purification by chromatography on arginine-Sepharose. Fraction volume, 1 ml.
Thus, due to a thorough purification from concomitant enzymes by differential centrifugation, we were able to isolate a highly active preparation of KEX2 proteinase, the yield being 74% of its initial total activity, without taking at least four additional chromatographic steps described in the literature (benzamidine-Sepharose, Con A-Sepharose, mono Q, and Superose 12) [1].Fig. 4. Electrophoresis of different protein preparations on 12% polyacrylamide gel plates: 1) crude membrane extract from sediment obtained by centrifugation at 80,000g for 20 min; 2) membrane extract obtained after differential centrifugation at rates from 7000g (15 min) to 20,000g (15 min); 3) active KEX2 fraction obtained by chromatography on DEAE-Servacel; 4) active KEX2 fraction obtained by chromatography on arginine-Sepharose; 5) protein standards.
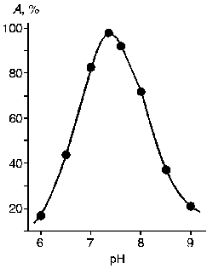
The purified KEX2 proteinase preparation was used to study the influence of pH (Fig. 5) and some inhibitors (HgCl2, PMS, LTI, STI, DFP, and EDTA) on its enzymatic activity. Human urine trypsin inhibitor (molecular weight 44,000 daltons) proved to be the most potent inhibitor, and so it was used in affinity chromatography as a ligand (Table 3).
TABLE 3. Influence of Inhibitors on KEX2 Proteinase ActivityFig. 5. Influence of pH on the activity of purified KEX2 proteinase toward Ac-Leu-Lys-Arg-pNA.

Pairs of basic amino acids are known to be the typical sites of proteolytic processing of biologically active peptides. The ability of KEX2 proteinase to participate in the processing of peptide hormones may be demonstrated with recombinant human proinsulin as an example. Proinsulin has two domains with potential sites of cleavage by KEX2 proteinase, i.e., -Arg31-Arg32v-Glu33- and -Lys67-Arg-68v-Gly69-. In rat pancreatic beta-cells, two KEX2-like proteinases have been found, each being specific toward one of the two cleavage sites of proinsulin, which is believed by the authors [24] to be the consequence of their cell localization. Significant discrepancies in data about proinsulin hydrolysis by KEX2-like proteinases from different sources [24-26] make it of interest to elucidate the specificity of the yeast KEX2 proteinase with respect to cleaving of this prohormone.
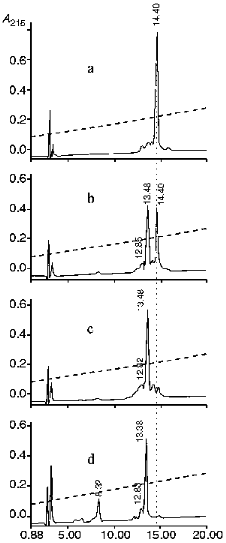
The analysis of N-terminal amino acids in reaction mixtures of proinsulin and KEX2 proteinase incubated at 37°C for different time periods showed that the N-terminal glutamine appears and its concentration increases in the course of the reaction, which is evidence that proinsulin is cleaved at the Arg32v-Glu33 site, as indicated. We determined the optimal ratio of proinsulin and KEX2 enzyme which ensures the complete conversion of proinsulin into its cleaved forms. Figure 6 shows the kinetics of KEX2 proteinase-mediated recombinant human proinsulin hydrolysis studied by HPLC. As seen from the chromatograms (a-c), at the 1:400 molar ratio of the enzyme and proinsulin complete disappearance of the proinsulin peak is observed 40 min after the initiation of the reaction, and, during this time period, another peak appears which represents the product of proinsulin hydrolysis. With 1:200 enzyme/proinsulin ratio, the same reaction proceed twice faster. It should be noted that, with KEX2 proteinase-mediated proinsulin cleavage, no C-peptide generation could be observed at any stage of the reaction. C-Peptide generation becomes possible when the products of hydrolysis are treated with trypsin (Fig. 6d). The reaction is significantly slower in ammonium-acetate buffer than in Tris-HCl buffer, the pH and proinsulin/enzyme ratio being the same.
It follows that KEX2 proteinase hydrolyzes only one of the two possible cleavage sites of proinsulin, i.e., -Arg31-Arg32v-Glu33-. The amino acid sequence on the right from the cleavage site of insulin, -Arg31-Arg32v-Glu33-Ala34-Glu35-Asp36-, resembles cleaved sequences -Lys-Argv-Glu-Ala-Glu-Ala- in pro-alpha-factor, the natural substrate of KEX2 proteinase [27]. There are no such analogs for amino acid sequences near the Lys67-Arg68 pair, and no hydrolysis of the pair by KEX2 proteinase was observed.Fig. 6. Reverse-phase HPLC of proinsulin (1.8 mg/ml) and the products of its enzymatic hydrolysis in Tris-HCl buffer (pH 7.0) at 37°C: a) proinsulin (elution time is 14.4 min); b) proinsulin after 20 min of exposure to KEX2 proteinase; c) proinsulin after 40 min of exposure to KEX2 proteinase (elution time of proteolytic product is 13.48 min); d) same as in (c) with trypsin added; exposure time is 20 min (elution times of C-peptide and cleavage product are 8.32 and 13.38 min, respectively). Abscissa, time of elution from Lichrosorb C-8 column (min); ordinate, optical density at 215 nm.
We have demonstrated that the purified KEX2 proteinase can cleave recombinant proteins in a specific manner. In particular, a hybrid protein, INF-link-HIV, which consists of the N-terminal fragment of gamma-interferon and a protease of human immunodeficiency virus connected by a linker sequence containing Lys-Arg, is hydrolyzed on the right from the C-end of this pair. The time course of the hydrolysis of the hybrid protein (within 1-22 and 2-15 h time periods) and the relationship between the process and the molar ratio of the enzyme and the recombinant substrate ((1) 1:500 and (3) 1:250) were studied using PAGE for monitoring the conversion of the protein into two fragments with lower molecular masses (Fig. 7). The complete hydrolysis of the recombinant protein at its ratio with the enzyme being 500:1 took 10-20 h, depending on temperature conditions. It should be noted that KEX2 proteinase cleaves the hybrid protein in a heterogenous medium yielding a homogenous set of products. Also, KEX2 proteinase has been shown to be functional in 4 M urea where it can hydrolyze the recombinant protein at high concentrations, up to 10 mg/ml, but the rate of hydrolysis decreases twofold. The sequencing of products of this reaction revealed the presence of the Pro-Gln-Ile sequence on the right from the cleaved bond, Lys81-Arg82v-, and Met-Gln-Asp-Pro (first amino acids in the gene-engineering construction) which confirms the suggestion about hydrolysis by Lys81-Arg82v- (Table 4). It is noteworthy that in the interferon fragment of the cleaved hybrid protein there is another pair of positively charged amino acids, Lys70-Arg71, but no hydrolysis of this pair was detected. Thus, in either the chimeric INF-link-HIV protein and recombinant proinsulin, KEX2 proteinase cleaves not every sequence that contains a pair of basic amino acids: this may be due to the specificity of the enzyme toward definite conformations of polypeptide domains near the cleaved bonds.
TABLE 4. Results of Sequencing of a Hybrid INF-link-HIV Protein Treated with KEX2 ProteinaseFig. 7. Electrophoresis in 16% polyacrylamide gel plates of a hybrid protein INF-link-HIV and products of its HEX-2 proteinase-mediated hydrolysis: 1) molar ratio of KEX2 and chimeric protein is 1:500; reaction time is 22 h; 2) molar ratio of KEX2 and chimeric protein is 1:500; reaction time is 15 h; 3) molar ratio of KEX2 and chimeric protein is 1:250; reaction time is 22 h; 4) the intact hybrid protein INF-link-HIV; 5) protein standards.

Thus, a highly active preparation of KEX2 proteinase thought to participate in peptide processing has been obtained. The proteinase has been partially characterized, and its ability to cleave recombinant proteins at the C-ends of Lys-Arg and Arg-Arg amino acid pairs has been shown to depend on the presence of certain amino acid sequences near the cleaved bonds.
The authors would like to thank N. I. Dergousova for providing recombinant INF-link-HIV protein and O. G. Ogloblina for samples of human urine trypsin inhibitor (HUTI) and Sepharose-immobilized HUTI.
LITERATURE CITED
1.Mizuno, K., Nakamura, T., Takada, K., Sakakibara,
S., and Matsuno, H. (1987) Biochem. Biophys. Res. Commun.,
144, 807-814.
2.Fuller, R. S., Brake, A., and Thorner, J. (1989)
Proc. Natl. Acad. Sci. USA, 86, 1434-1438.
3.Thomas, G., Thorne, B., and Thomas, L. (1988)
Science, 241, 226-230.
4.Brennan, S. D., and Peach, R. G. (1988) FEBS
Lett., 229, 167-170.
5.Achstetter, T., and Wolf, D. H. (1985) EMBO
J., 4, 173-177.
6.Fuller, R. S., Sterne, R. E., and Thorner, J.
(1988) Annu. Rev. Physiol., 50, 345-362.
7.Jean, F., Boudreault, A., Basak, A., Seidah, N. G.,
and Lazure, C. (1995) J. Biol. Chem., 270,
13277-13284.
8.Lipkind, G., Gong, Q. M., and Steiner, D. F. (1995)
J. Biol. Chem., 270, 13277-13284.
9.Ledgerwood, E. C., Brennan, S. O., Cawley, N. X.,
Loh, Y. P., and George, P. M. (1996) FEBS Lett., 383,
67-71.
10.Ledgerwood, E. C., George, P. M., Peach, R. J.,
and Brennan, S. O. (1995) Biochem. J., 308, 321-325.
11.Zsebo, K. M., Lu, H.-S., and Fieschko, J. C.
(1986) J. Biol. Chem., 261, 5858-5865.
12.Bitter, G. A., Chen, K. K., and Banks, A. R.
(1984) Proc. Natl. Acad. Sci. USA, 81, 5330-5334.
13.Brake, A. J., Merryweather, J. P., and Coit, D.
G. (1984) Proc. Natl. Acad. Sci. USA, 81, 4642-4646.
14.Bradford, M. (1976) Anal. Biochem.,
72, 248-254.
15.Laemmli, U. K. (1970) Nature, 27,
680-685.
16.Mizuno, K., Nakamura, T., Ohshima, T., Tanaka,
S., and Matsuo, H. (1989) Biochem. Biophys. Res. Commun.,
159, 305-311.
17.Mizuno, K., Nakamura, T., and Matsuo, H. (1989)
Biochem. Biophys. Res. Commun., 164, 780-787.
18.Redding, K., Holcomb, C., and Fuller, K. S.
(1991) J. Cell Biol., 13, 527-539.
19.Redding, K., Seeger, M., Payne, G. S., and
Fuller, K. S. (1996) Mol. Biol. Cell, 7, 1667-1977.
20.Vinogradov, A. D. (ed.) (1990) Biological
Membranes. Methods [Russian translation], Mir, Moscow, p. 68.
21.Wachter, E., Deppner, K., and Hochstrasser, K.
(1980) FEBS Lett., 119, 58-62.
22.Ogloblina, O. G. (1982) Biokhimiya,
47, 1587-1600.
23.Ogloblina, O. G., and Aref'eva, T. I. (1994)
Biochemistry (Moscow), 59, 340-352 (Russ.).
24.Rhodes, C. J., Lincoln, B., and Shoelson, S. E.
(1992) J. Biol. Chem., 267, 22719-22727.
25.Thim, L., Hansen, M. T., and Norris, K. (1986)
Proc. Natl. Acad. Sci. USA, 83, 6766-6770.
26.Fuller, R. S., Brake, A. J., and Thorner, J.
(1989) Science, 246, 482-486.
27.Kurjan, J., and Herskowitz, I. (1982)
Cell, 30, 933-943.