Site-Specific Endonucleases RspLKI and RspLKII from Rhodococcus species LK2 are Isoschizomers of SphI and BamHI
E. V. Zabaznaya,1 L. A. Zheleznaya,2 and N. I. Matvienko1,3
1Institute of Protein Research, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia; fax: (095) 924-0493; E-mail: protres@sovam.com2Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia; fax: (095) 135-6219.
3To whom correspondence should be addressed.
Submitted February 25, 1997; revision submitted April 3, 1997.
Two site-specific endonucleases, RspLKI and RspLKII, have been isolated and purified to functional homogeneity from the soil bacterium Rhodococcus species LK2. RspLKI recognizes the 5´-GCATGvC-3´ DNA sequence and RspLKII recognizes the 5´-GvGATCC-3´ sequence (arrows indicate DNA cleavage sites). The isolated enzymes are class II site specific endonucleases and are isoschizomers of endonucleases SphI and BamHI, respectively.
KEY WORDS: site-specific endonuclease, isoschizomer, phosphocellulose, heparin-Sepharose, hydroxyapatite.
More than 2000 site-specific endonucleases (EC 3.1.21.4) are identified now and they recognize about 200 different sites on DNA [1, 2]. The search for new enzymes continues. The goals of these studies are both isolation of new endonucleases specific with respect to new sequences and detection of isoschizomers differing from prototypes by higher content in a cell, stability during storage, low non-specific activity, and allowing simplified purification of the enzyme.
Here we describe isolation and characterization of endonucleases RspLKI and RspLKII from soil strain Rhodococcus species LK2. These enzymes are isoschizomers of SphI and BamHI.
MATERIALS AND METHODS
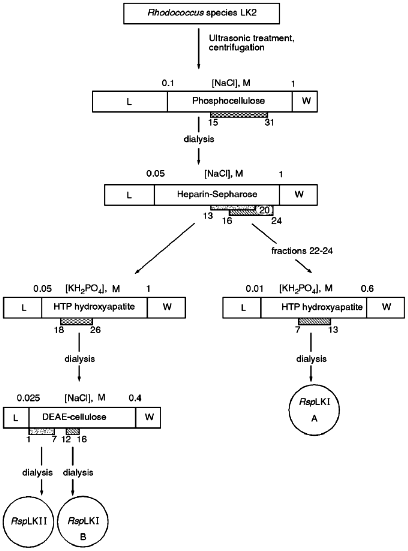
Isolation and Purification of Endonucleases. Strain Rhodococcus species LK2 isolated from soil was used in the study. The culture was grown at 30°C in 2 liters of intensively aerated LB medium [3] to late logarithmic phase. Cells were sedimented by centrifugation and biomass (18 g) was resuspended in 79 ml of buffer A (10 mM KH2PO4, pH 7.0, 1 mM EDTA, 0.1 M NaCl, 7 mM mercaptoethanol) containing 1 mM phenylmethylsulfonyl fluoride. Cells were lysed in an ice bath using a UZDN-1A ultrasonic disintegrator for 28 min (60 sec pulses with 60 sec intervals). Disintegration was monitored by the decrease in turbidity of the cellular suspension at 540 nm in a KFK-2-UKhL 4.2 photoelectrocolorimeter. The lysate was centrifuged for 30 min at 25,000 rpm in a Ti50 rotor (Beckman, USA). The resulting supernatant (75 ml) was applied at the rate 20 ml/h onto a phosphocellulose P-11 column (Whatman, England) equilibrated with buffer A. The sorbent volume was 30 ml. The column was washed with buffer A and bound proteins were eluted by a linear gradient of NaCl (0.1-1.0 M) in buffer B (10 mM KH2PO4, pH 7.0, 1 mM EDTA, 7 mM 2-mercaptoethanol) at the flow rate 9 ml/h. Total gradient volume was 180 ml and the time of collection of each fraction was 30 min. After the gradient the column was washed with 30 ml of 1 M NaCl in buffer B. Fractions of the gradient possessing the enzyme activity were dialyzed against buffer B containing 50 mM NaCl and then were applied onto a column with heparin-Sepharose (Pharmacia, Sweden) equilibrated with buffer B containing 50 mM NaCl. The sorbent volume was 7 ml, and the rate of application was 20 ml/h. Proteins bound to the sorbent were eluted with 40 ml of a linear gradient of NaCl (0.05-1.0 M) in buffer B at flow rate 20 ml/h. The time of collection of each fraction was 6 min. After the gradient the column was washed with 22 ml of 1 M NaCl in buffer B. Fractions 22-24 exhibiting endonuclease activity (Fig. 1) were pooled, diluted 10-fold with buffer C (10 mM KH2PO4, pH 7.0, 7 mM 2-mercaptoethanol), and applied onto HTP hydroxyapatite (Bio-Rad, USA) equilibrated with the same buffer. The sorbent volume was 2.5 ml and the application rate was 8 ml/h. After column washing with 30 ml of buffer C, proteins bound to the sorbent were eluted with a linear gradient (0.01-0.6 M) of potassium phosphate buffer, pH 7.0, containing 7 mM 2-mercaptoethanol. The gradient volume was 40 ml, the elution rate was 20 ml/h, and a time for collection of each fraction was 8 min. Fractions exhibiting endonuclease activity were pooled and after the addition of serum albumin (molecular biology grade, Olaine, Latvia) to final concentration 50 µg/ml were initially dialyzed overnight at 4°C against buffer D (10 mM Tris-HCl, pH 7.5, 50 mM KCl, 0.1 mM EDTA, 7 mM 2-mercaptoethanol). Then under the same conditions they were dialyzed against the storage buffer (10 mM Tris-HCl, pH 7.5, 100 mM KCl, 0.1 mM EDTA, 0.1 mM dithiothreitol, 50% glycerol).
Fractions 13-21 after heparin-Sepharose exhibiting mixed endonuclease activity were pooled and applied onto a HTP hydroxyapatite column equilibrated with buffer C. The sorbent volume was 25 ml, application rate was 17 ml/h, and the time for collection of each fraction was 3 min. The column was washed with 25 ml of buffer C and bound proteins were eluted by a linear gradient (0.05-1.0 M) of potassium phosphate buffer, pH 7.0, containing 7 mM 2-mercaptoethanol. Fractions demonstrating mixed endonuclease activity were dialyzed against buffer E (10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 7 mM 2-mercaptoethanol) containing 25 mM NaCl and then were applied onto a DEAE-Tris-acryl column (Pharmacia) equilibrated with buffer E containing 25 mM NaCl. The sorbent volume was 7 ml and the application rate was 40 ml/h. The column was washed with 23 ml of buffer E containing 25 mM NaCl and bound proteins were eluted with a linear gradient of NaCl (0.025-0.4 M) in buffer E at flow rate 15 ml/h. Time of collection of each fraction was 10 min. Fractions demonstrating various endonuclease activities were pooled and after addition of albumin were dialyzed against storage buffer as described above.Fig. 1. General scheme for isolation of site-specific endonucleases RspLKI and RspLKII. Rectangles in the scale indicate gradients of eluting buffers. Initial and final molarities are indicated above them. Numbers of gradient fractions are given below them. L corresponds to the step of application and washing; W corresponds to column wash after the gradient. Hatched rectangles correspond to fractions exhibiting endonuclease activity, and circles show final enzyme preparations.
Determination of Endonuclease Activity. Activities of the isolated enzymes were assayed in 40 µl of MRB buffer (10 mM Tris-HCl, pH 8.0, 10 mM MgCl2) containing 0.3 µg of phage lambda DNA (lambdaCI85757, dam-, dcm-) and 2 µl of enzyme preparation from fractions or diluted preparations. The mixture was incubated for 1 h at 37°C and then reaction products were analyzed by electrophoresis in 1% agarose gel. One unit of the activity corresponded to amount of enzyme required for complete hydrolysis of 1 µg of phage lambda DNA in 1 h at 37°C in MRB buffer in 40 µl of the reaction mixture.
Determination of a Functional Purity of the Enzymes. The absence of nuclease contamination was determined by the method of restriction-ligation-restriction [4].
Determination of Points of DNA Cleavage. Phages M13tg130 and M13tg131 were used to determine the points of DNA cleavage. Cleavage points were detected by the elongated primer method [5].
RESULTS AND DISCUSSION
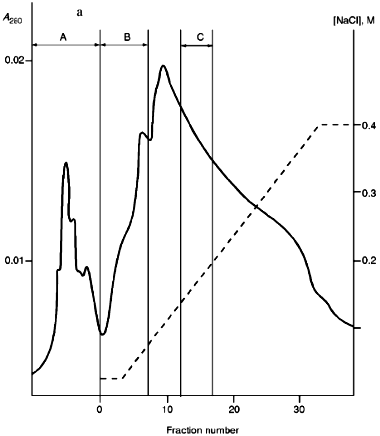
Isolation of Site-Specific Endonucleases. Primary screening of a strain Rhodococcus species LK2 for the presence of site-specific endonucleases revealed restrictase RspLKI. Preliminary data suggested that it is an isoschizomer of restrictase SphI. However, during isolation of this enzyme we found another restrictase, RspLKII, which we did not detect earlier due to its low endonuclease activity missed on the background of high activity of RspLKI. Thus, we had to correct our plans not only of isolation of functionally pure enzyme, but also of separation of two activities. Figure 1 shows the general scheme of the purification procedure. A phosphocellulose column was used as the first column. The use of this column permits excluding a step of nucleic acid precipitation from the lysate and to remove oligonucleotides, non-specific endonucleases, and acid proteins [6]. However, the phosphocellulose step did not separate the restrictases (Fig. 2, a and b). Pooled fractions 15-31 were dialyzed for reduction of NaCl and applied onto a heparin-Sepharose column. Elution by a linear gradient of NaCl did not result in complete separation of the enzymes. Endonuclease RspLKI was eluted at 0.56-0.95 M NaCl whereas RspLKII was eluted at 0.44-0.77 M NaCl (Fig. 3, a and b). Fractions 22-24 containing enzyme RspLKI were pooled and after dilution they were applied onto HTP hydroxyapatite. Figure 4 shows an optical elution profile and analysis of endonuclease activity of RspLKI enzyme in gradient fractions. The zone of endonuclease elution corresponds to 0.14-0.3 M (Fig. 4a), and endonuclease activity was detected in fractions 6-26 (Fig. 4b). Peak fractions 7-13 (Fig. 4c) were pooled and after addition of albumin were transferred into the storage buffer.
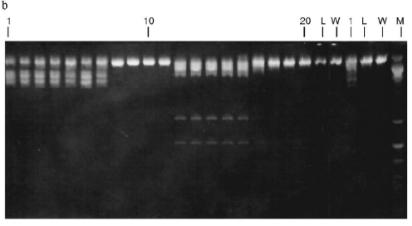
Fig. 2. a) Optical elution profile of the phosphocellulose column. Solid line shows A280; the dashed line indicates the NaCl molarity in the gradient fractions; A, application fraction; B, washing fraction; C, fractions containing restrictase activity; D, column wash with high salt concentration. b) Analysis of the activity of site-specific endonucleases in gradient fractions from the phosphocellulose column. Numbers indicate fraction numbers; L indicates fractions of application on the column and washings; W is a fraction of a column wash with high salt concentration. For incubation with phage lambda DNA the fractions were diluted 1:10.
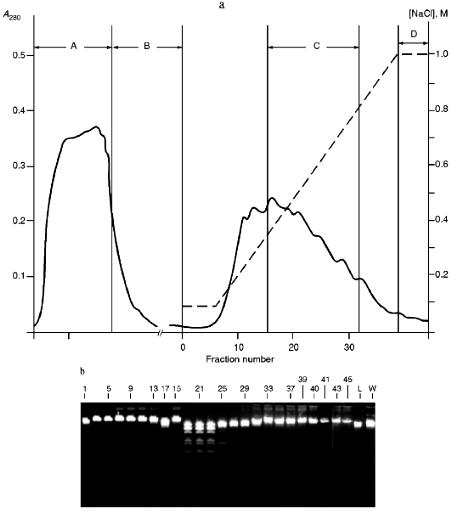
Fig. 3. a) Optical elution profile of the heparin-Sepharose column. The solid line shows A280; dashed line indicates NaCl molarity in the gradient fractions; A, fractions containing RspLKII; B, fractions containing RspLKI; C, column wash with high salt concentration. b) Analysis of endonuclease activity in gradient fractions from the heparin-Sepharose column. L indicates fractions of application on the column and washings; W is a fraction of a column wash with high salt concentration. Numbers indicate fraction numbers. For the incubation with phage lambda DNA the fractions were diluted 1:20.
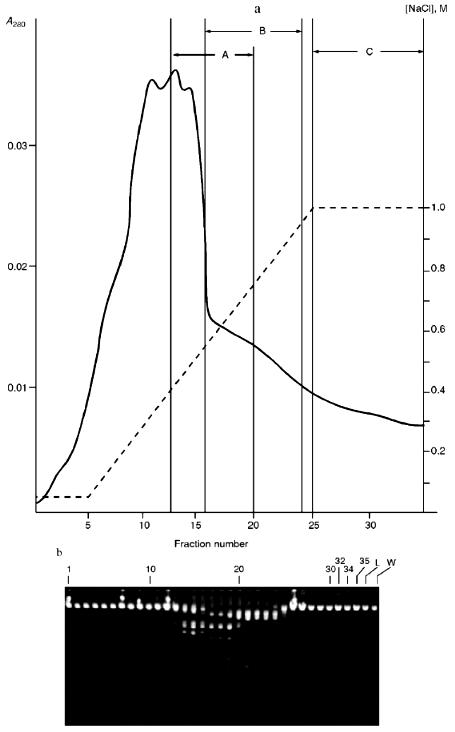
Fractions 13-21 after heparin-Sepharose were used for subsequent enzyme separation. Attempts to separate the restrictases on a hydroxyapatite column were not successful (Fig. 5, a and b). Satisfactory separation of proteins was achieved using DEAE-cellulose. Figure 6 shows the optical elution profile and analysis of endonuclease activity of the isolated enzymes in gradient fractions. The zone of endonuclease RspLKI elution corresponds to 0.025-0.075 M NaCl; restrictase activity appears in fractions 1-7. The zone of R RspLKII elution corresponds to 0.14-0.19 M and the enzyme activity was detected in fractions 12-16. Fractions 1-7 and 12-16 were pooled and after addition of albumin were transferred into the storage buffer.Fig. 4. a) Optical elution profile of the hydroxyapatite column. A and B, fractions of application and wash; C, fractions containing restrictase RspLKI; D, column wash with high salt concentration. Solid line shows A280; dashed line indicates KH2PO4 molarity. b) Analysis of endonuclease activity RspLKI in gradient fractions from HTP hydroxyapatite column. L and L1 are fractions of application and washing; W is a fraction of column washing with high salt concentration. Numbers indicate fraction numbers. The fractions were not diluted for incubation with phage lambda DNA. M is a marker of fragment lengths of lambda:IS4. c) Analysis of RspLKI activities in gradient fractions from hydroxyapatite HTP column. For the incubation with phage lambda DNA the fractions were diluted 1:10.
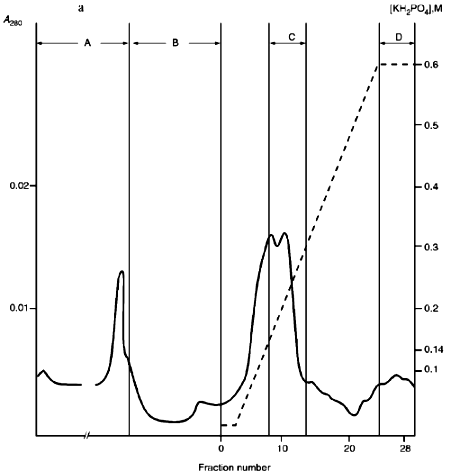
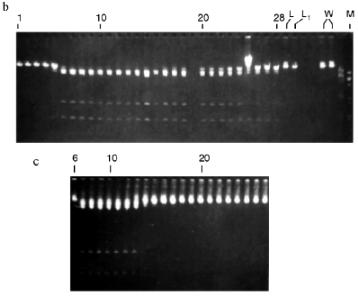
Fig. 5. a) Optical elution profile of the hydroxyapatite column of fractions 13-21 after heparin-Sepharose. A and B, fractions of application and wash; C, fractions exhibiting mixed endonuclease activity. b) Analysis of endonuclease activity in gradient fractions from HTP hydroxyapatite column. L, L1, and W are fractions of application, washing, and column washing with high salt concentration, respectively. For incubation with phage lambda DNA the fractions were diluted 1:10. M is a marker of fragment lengths (lambda:IS4).
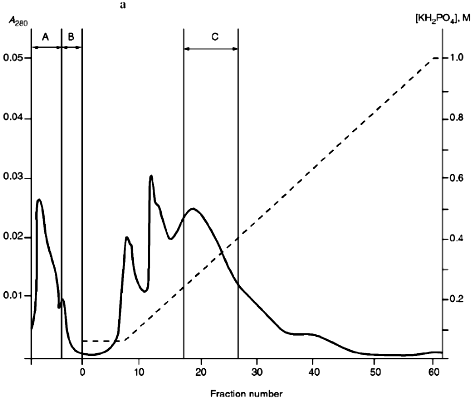
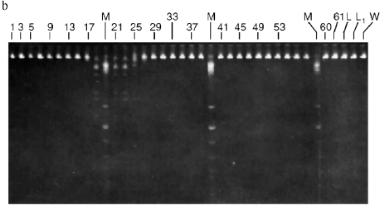
Total activities of isolated preparations of the enzyme RspLKI, Ra and Rb (Fig. 1), were 150,000 and 16,000 units with phage lambda DNA as the substrate. This corresponds to a total activity of 166,000 units. The total activity of the enzyme RspLKII was 18,500 units. The yield of RspLKI and RspLKII was 9000 and 1000 units per g of wet biomass, respectively.Fig. 6. a) Optical elution profile of the DEAE-cellulose column. A, fractions of application and washing; B, fractions containing endonuclease RspLKII; C, fractions containing RspLKI. b) Analysis of enzyme activities in gradient fractions from DEAE-cellulose column. The fractions were not diluted for incubation with phage lambda DNA. L, fractions of application and washing; W, fractions of column washing with high salt concentration.
The functional purity of the enzyme preparations was confirmed by both reactions of restriction-ligation-restriction (Fig. 7, a and b) and applicability of these enzymes for sequencing during determination of DNA cleavage points.
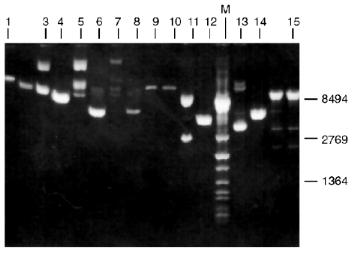
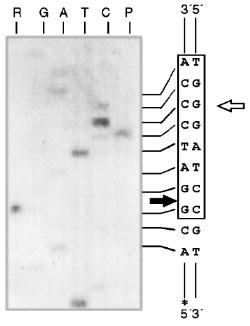
Determination of Recognition Sites and Cleavage Points. Phage T7 DNA is not a substrate of endonuclease RspLKI. The enzyme catalyzes cleavage of phage lambda DNA at 6 points resulting in the appearance of fragments with various lengths of 12044, 11940, 9790, 9084, 3003, 2212, and 429. Figure 8 shows fragments 3003 and 2212; high-molecular-weight fragments 12044, 11940, 9790, and 9084 are distributed in the gel as a single wide band. Fragment with length 429 is invisible under these conditions of electrophoresis. This restrictase catalyzes a single point cleavage of DNA of plasmids pBR322, pACYC184, and pJRD184 and phage M13tg131 (Fig. 8). This cleavage behavior is similar to that of restrictase SphI so it was suggested that RspLKI is an isoschizomer of SphI. This was confirmed during determination of DNA cleavage points. Figure 9 shows that the enzyme RspLKI recognizes the palindrome sequence 5´-GCATGvC-3´ and cleaves it as the arrow indicates. So the enzyme is an isoschizomer of SphI isolated from Streptomyces phaeochromogenes. The yield of the enzyme SphI was 3,000 units per g of biomass [7]. The yield of RspLKI is 9,000 units per g of biomass with phage lambda DNA as a substrate. Thus, Rhodococcus species LK2 exceeds threefold the strain Streptomyces phaeochromogenes by the yield of the enzyme.Fig. 7. Reaction of restriction-ligation-restriction. a: 1) Phage lambda DNA; 2) phage lambda DNA cleaved by RspLKI (Ra); 3) ligated phage lambda DNA; 4) repeated restriction of the ligated DNA by RspLKI (Ra); 5) phage lambda DNA cleaved by RspLKI (Rb); 6) ligated phage lambda DNA; 7) repeated restriction of ligated DNA by RspLKI (Rb). M is a marker of fragment lengths (lambda:IS4). b: 1) Phage lambda DNA cleaved by RspLKII; 2) ligated phage lambda DNA; 3) repeated restriction of ligated DNA by RspLKII; 4) phage lambda DNA. M is a marker of fragment lengths.
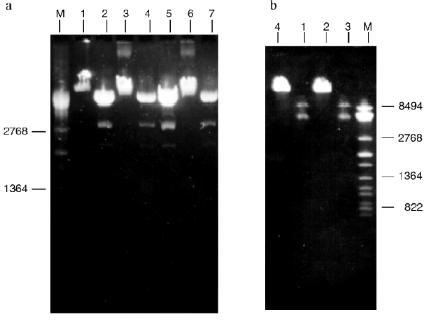
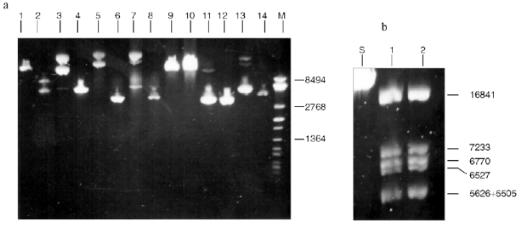
Fig. 8. Cleavage of various substrates by RspLKI (Ra). Lanes: 1, 15) phage lambda DNA; 3, 4) phage M13tg131 DNA; 5, 6) plasmid pACYC184 DNA; 7, 8) plasmid pBR322 DNA; 9, 10) phage T7 DNA; 11, 12) plasmid pJRD184 DNA; M is a marker of fragment lengths (lambda:IS4); 13, 14) plasmid pUC19 DNA. Lanes 4, 6, 8, 10, 12, 14, and 15 demonstrate substrate DNA cleaved by RspLKI. Lanes 1, 3, 5, 7, 9, 11, and 13 demonstrate uncleaved substrate DNA.
Endonuclease RspLKII does not cleave phage T7 DNA and does not catalyze a single point cleavage of DNA of plasmids pBR322, pACYC184, and pJRD184 and phage M13tg131 (Fig. 10a). The enzyme RspLKII has five cleavage sites on phage lambda DNA that results in formation of fragments of various lengths: 16841, 7233, 6770, 6527, 5626, and 5505. Satisfactory resolution of these fragments can be achieved during electrophoresis in 0.7% gel. Figure 10b shows fragments 16841, 7233, 6770, and 6527. Fragments 5626 and 5505 are present as a single band in the gel. Figure 11 shows that RspLKII recognizes the sequence 5´-GvGATCC-3´ and cleaves it as the arrow indicates, i.e., the enzyme is isoschizomer of BamHI. Methylation of phage lambda DNA by dam methylase at sequences GATC does not influence the rate of its hydrolysis catalyzed by endonuclease RspLKII. Thus, the enzyme RspLKII, like BamHI, is insensitive to dam-methylation.Fig. 9. Determination of cleavage points of DNA by endonuclease RspLKI (Ra). T, G, A, and C are sequencing lanes; R is a product of polymerase reaction cleaved by RspLKI; P is the same but after DNA completion with Klenow fragment.
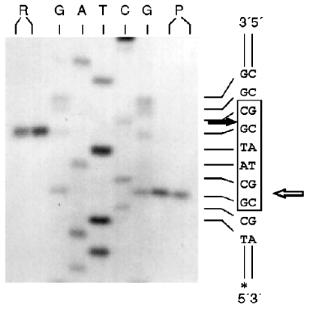
Fig. 10. a) Cleavage of various substrates by RspLKII. Lanes: 1, 2) phage lambda DNA; 3, 4) phage M13tg131 DNA; 5, 6) plasmid pACYC184 DNA; 7, 8) plasmid pBR322 DNA; 9, 10) phage T7 DNA; 11, 12) plasmid pJRD184 DNA; 13, 14) plasmid pUC19 DNA. M is a marker of fragment lengths (lambda:IS4). Lanes 2, 4, 6, 8, 10, 12, and 14 demonstrate substrate DNA cleaved by RspLKII. Lanes 1, 3, 5, 7, 9, 11, and 13 demonstrate uncleaved substrate DNA. b) Cleavage of phage lambda DNA by RspLKII. Lanes: S, uncleaved phage lambda DNA; 1) phage lambda DNA cleaved by RspLKII; 2) phage lambda DNA cleaved by enzyme BamHI (Fermentas, Lithuania). Products of the cleavage were analyzed in 0.7% agarose gel.
Some Properties of Restrictases RspLKI and RspLKII. For elucidation of optimal conditions for the activity of the isolated enzymes we varied ionic strength, pH of the buffer, and incubation temperature. The enzyme stability at room temperature, alteration of enzyme activity during long-term incubation, and thermoinactivation of the enzymes were also studied.Fig. 11. Determination of cleavage points of DNA by endonuclease RspLKII. T, G, A, and C are sequencing lanes; R is a product of the polymerase reaction cleaved by RspLKII; P is the same but after DNA completion with Klenow fragment.
Change of NaCl concentration in the reaction buffer (10 mM Tris-HCl, pH 7.4, 200 µg/ml albumin, 10 mM MgCl2) influenced enzyme activity. The activity of RspLKI (Rb) was fourfold higher in the buffer containing 50 mM NaCl (MRB) [3] than in the buffer without NaCl (LRB), whereas in the buffer containing 100 mM NaCl (HRB) the enzyme was inactive. The activity of RspLKII was more than threefold higher in MRB buffer compared with LRB and HRB.
The optimum pH for both enzymes is 7.4. The use of more alkaline buffers (pH 8.0, 8.5, and 9.0) resulted in a twofold decrease in enzyme activities.
The highest rate of phage lambda DNA hydrolysis by RspLKI (Rb) was observed at 37°C. Decrease of the incubation temperature to 30°C and increase of the temperature to 48°C caused a twofold decrease in the enzyme activity. The maximal rate of phage lambda DNA cleavage by endonuclease RspLKII was observed at 22°C. Increase of the temperature to 37 and 48°C decreased enzyme activity by 1.6- and 2-fold, respectively. At 55°C the enzyme is inactive.
Storage of the enzyme preparations during 3 months at -10°C did not alter their activities. Storage at 20°C during 2 days decreased activity of RspLKI and RspLKII by 1.6- and 2.5-fold, respectively.
Comparison of phage lambda DNA cleavage patterns during 1 and 16 h incubation at 37°C revealed that total cleavage of substrate DNA by both RspLKI and RspLKII requires twofold less enzyme for overnight incubation compared with 1 h incubation. After preheating the enzyme for 20 min at 65°C in the optimal buffer containing pBR322 DNA, hydrolysis of subsequently added phage lambda DNA was not observed. Thus, both enzymes can be inactivated after hydrolysis by short-term heating of the incubation mixture.
This work was supported by the Russian Academy of Sciences and Russian Foundation for Basic Research (grant No. 96-04-49050).
LITERATURE CITED
1.Roberts, R. J., and Macelis, D. (1992) Nucleic
Acids Res., 20, Suppl., 2167-2180.
2.Nelson, M., and McClelland, M. (1991) Nucleic
Acids Res., 19, Suppl., 2071-2145.
3.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor Lab. Press, Cold Spring Harbor.
4.New England Biolabs Catalog (1996/1997) New
England Biolabs Inc., Beverly.
5.Brown, N. L., and Smith, M. (1980) Meth.
Enzymol., 65, 391-404.
6.Green, P. J., Heyneker, H. L., Bolivar, F.,
Rodriquez, R. L., Betlach, M. C., Covarrubias, A. A., Backman, K.,
Russel, D. J., Tait, R., and Boyer, H. W. (1978) Nucleic Acids
Res., 5, 2373-2380.
7.Fuchs, L. Y., Covarrubias, L., Escalante, L.,
Sancherr, S., and Bolivar, F. (1980) Gene, 10,
39-46.