Site-Specific Endonuclease from Thermophilic Bacillus Species MK Strain is Isoschizomers of SalI
M. A. Kerzhner,1 S. A. Shiryaev,1 L. A. Zheleznaya,2 and N. I. Matvienko1,3
1Institute of Protein Research, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia; fax: (095) 924-0493; E-mail: protres@sovam.com2Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia; fax: (095) 135-6219.
3To whom correspondence should be addressed.
Submitted February 25, 1997; revision submitted April 3, 1997.
Screening of thermophilic bacterial strains revealed a strain containing site-specific endonuclease BspMKI. This endonuclease was purified to functional homogeneity during sequential chromatographic steps. The enzyme recognizes sequence 5´-GvTCGAC-3´ on DNA molecule and is isoschizomer of endonuclease SalI. The molecular mass of BspMKI is about 45 kD. The enzyme is maximally active at 55°C and MRB (50 mM NaCl, 10 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 1 mM dithiothreitol) is the optimal buffer. The enzyme is highly stable and retains its activity during two weeks at room temperature.
KEY WORDS: site-specific endonuclease, isoschizomer, restriction, sequencing.
More than 2000 site-specific endonucleases recognizing more than 200 specific sequences are identified now [1]. The search for new enzymes continues. Site-specific endonucleases possessing higher stability compared with known enzymes are of interest. Site-specific endonucleases type II (EC 3.1.21.4) recognizing palindrome sequences are used particularly often. Thus, identification of new site-specific endonucleases of this type or isomers of known endonucleases possessing valuable new properties opens wider possibilities in gene engineering.
Here we describe the isolation of a site-specific endonuclease from the thermophilic strain Bacillus species MK. The paper also describes methods of the enzyme isolation, determination of cleavage points of the recognized sequence and molecular mass, and also some other properties.
MATERIALS AND METHODS
Substrate DNAs used in this study were isolated in the group of molecular genetics (Institute of Protein Research, Russian Academy of Sciences).
Isolation of Site-Specific Endonuclease. A strain of the thermophilic soil bacterium Bacillus species MKI was used for enzyme isolation. The culture was grown at 55°C using LB medium (10 g tryptone, 5 g yeast extract, 10 g NaCl per liter of medium) to late logarithmic phase of the growth under intensive aeration. Biomass was concentrated by centrifugation. The sediment was washed with TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA), centrifuged again, and kept at -20°C. The yield of biomass was 19 g from 4 liters of the medium. For disruption of cells biomass (9 g) thawed at room temperature was resuspended in 30 ml of lysing buffer (20 mM Tris-HCl, pH 8.0, 10 mM EDTA, 7 mM 2-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride). After the addition of 5 ml of lysozyme solution (30 mg/ml) freshly prepared in the same buffer, the suspension was incubated for 15 min in an ice bath. Cells were lysed in the ice bath using a UZDN-1A ultrasonic disintegrator for 10 min (30 sec pulses with 30 sec intervals). The degree of cell disruption was monitored at 590 nm using a KFK-2-UKhL 4.2 nephelometer. A tenfold decrease of turbidity of the suspension indicates good cell disruption. Cellular fragments were sedimented by centrifugation at 12,000 rpm for 30 min using a K-24 Janetzki centrifuge (Germany). For the isolation of nucleic acids KCl was added to suspension to the final concentration of 250 mM and then 13 ml of 5% solution of Polyimine P (BRL, USA) were gradually added during 1 h with stirring. The solution was centrifuged in a Beckman centrifuge for 30 min at 25,000 rpm (rotor Ti45). Ammonium sulfate was added to the supernatant to 70% saturation and after its complete dissolution the mixture was incubated 20 min with stirring and centrifuged 30 min at 35,000 rpm (rotor Ti45). The sediment was resuspended in 20 ml of buffer A (10 mM Tris-HCl, pH 7.4, 7 mM 2-mercaptoethanol) containing 1 mM EDTA and 100 mM NaCl and applied onto a Sephadex G-100 (Pharmacia, Sweden) column (80 × 5 cm, volume 1700 ml) equilibrated with buffer A. Fractions of 30 ml were collected at a flow rate of 40 ml/h. The optical elution profile was determined by absorbance at 280 nm in a flow cuvette in a UV-1 spectrophotometer (Pharmacia). Fractions containing site-specific endonucleases were pooled and applied at flow rate 8 ml/h onto a Blue Sepharose column (Pharmacia) equilibrated with buffer A containing 100 mM NaCl. The sorbent volume was 13 ml. Elution was carried out at flow rate 15 ml/h using 130 ml of buffer A containing a linear gradient of NaCl (0.1-2.5 M). Fractions possessing the enzyme activity were pooled (volume 27.5 ml) and dialyzed against buffer A containing 0.05 M NaCl. The resulting enzyme preparation was applied at flow rate 12 ml/h onto a heparin-Sepharose column (volume 8 ml) equilibrated with buffer A containing 0.05 M NaCl. It was eluted by buffer A containing a gradient of NaCl (0.05-1.0 M). The total volume was 80 ml. Fractions containing endonuclease (volume 22 ml) were pooled and after addition of serum albumin of "molecular biology grade" (Olaine, Latvia) to 100 µg/ml were dialyzed against storage buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol, and 50% glycerol).
Determination of Endonuclease Activity in Fractions. Aliquots of 3-5 µl from fractions were incubated with 25 µl of MRB buffer (10 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 50 mM NaCl) containing 0.5 µg of plasmid pUC19 DNA for 1 h at 55°C. Reaction products of DNA cleavage were analyzed by electrophoresis in 1% agarose gel.
Molecular mass of BspMKI was determined by gel chromatography on a Sephacryl S200HR column (volume 36 ml) in 20 mM Tris-HCl buffer, pH 8.0, containing 200 mM NaCl, 1 mM DTT, and 1 mM EDTA. The column was precalibrated with marker proteins: 1) albumin (68 kD); 2) ovalbumin (43 kD); 3) trypsin inhibitor (20 kD). The flow rate of the elution was 8 ml/h. The yield of the calibration proteins was monitored in a UV-1 flow spectrophotometer and endonuclease BspMKI was detected by
enzymatic activity in fractions.
Cleavage points on DNA were determined by the elongated primer method [2]. Phage M13tg130 DNA was used as a matrix [3]. It contains recognition site for BspMKI near the universal primer.
RESULTS
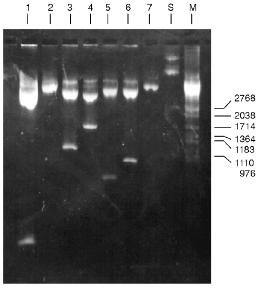
Isolation of Site-Specific Endonucleases. Screening of a collection of microorganism strains for the presence of site-specific endonucleases revealed a site-specific endonuclease in the strain Bacillus species MK. For investigation of its properties, the enzyme was purified to functional homogeneity using sequential chromatographies on various sorbents. Figure 1 shows the general scheme of the purification of endonuclease BspMKI. Blue Sepharose and heparin-Sepharose are traditional sorbents used for purification of site-specific endonucleases [4]. However, application of the extract without removal of nucleic acids by polyethylenimine (PEI) resulted in a significant part of the endonuclease appearing in the application and washing fractions. To avoid this, DNA was sedimented by PEI in the presence of 250 mM KCl which prevents DNA binding to enzymes. Proteins were sedimented from the solution containing PEI by ammonium sulfate (70% saturation) with subsequent desalting on a Sephadex G-100 column. Fractions 11-16 containing endonuclease (Fig. 2) were pooled and applied onto a Blue Sepharose column. Figure 3a shows the optical elution profile from a gradient Blue Sepharose column, and Fig. 3b shows analysis of the endonuclease activity in the gradient fractions. The zone of BspMKI elution from the column is wide, but only fractions exhibiting maximal activity (total hydrolysis of substrate DNA) were used in the subsequent purification steps. Fractions from 4 to 8 eluting at 0.40-0.70 M NaCl were pooled and dialyzed against buffer A containing 0.05 M NaCl. The resulting preparation was applied onto a heparin-Sepharose column. The zone of restrictase elution corresponds to 0.25-0.40 M NaCl (Fig. 4). Fractions containing enzyme activity were pooled and after the addition of serum albumin to final concentration 100 µg/ml were dialyzed overnight against storage buffer. The yield of the enzyme was 70,000 units from 9 g of biomass. The resulting preparation was purified to functional homogeneity. This was confirmed by the restriction--ligation--restriction test (Fig. 5). The cleavage pattern of the initial plasmid (lane 2) was identical to that observed after repeated cleavage after ligation (lane 4). Possible presence of nonspecific endonuclease activity would not allow the ligation of the cleaved plasmid (lane 3). Satisfactory results of determination of cleavage points on DNAdetermined by the elongated primer method [2] also confirm the purity of the enzyme preparation. This method requires pure enzyme free of endonuclease and phosphatase activities.
Fig. 1. General scheme of isolation of site-specific endonuclease BspMKI from the strain Bacillus species MK.
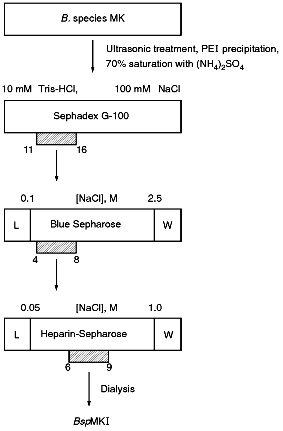
Fig. 2. Purification of the endonuclease BspMKI on a Sephadex G-100 column: a) optical elution profile (A280) of the column. A, fractions containing BspMKI; b) analysis of the fractions on endonuclease activity. S, substrate pUC19 DNA without cleavage; pUC19 DNA treated with 5 µl aliquots from fractions (numbers indicate fraction numbers). M indicates fragments lengths (phage lambda DNA cleaved by BbvII), fragment sizes (in nucleotide pairs) are indicated on the right.
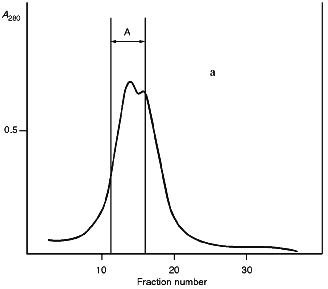
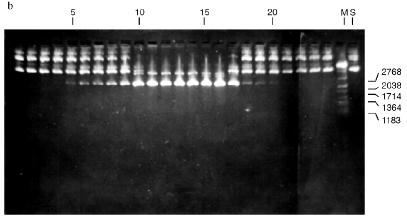
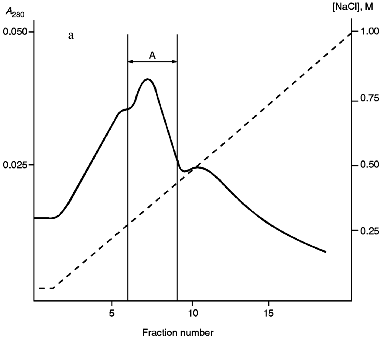
Fig. 3. Purification of endonuclease BspMKI on Blue Sepharose column: a) optical elution profile (A280) of column. Solid line shows optical density; dashed line indicates NaCl molarity in the gradient fractions. A, fractions containing BspMKI; b) analysis of endonuclease activity in gradient fractions. S, substrate pUC19 DNA without cleavage; pUC19 DNA treated with 5 µl aliquots from applied fraction (L1), washing fraction (L2), and elution fractions (numbers indicate fraction numbers). M indicates fragments lengths (phage lambda DNA cleaved by BbvII), fragment sizes (in nucleotide pairs) are indicated on the right.
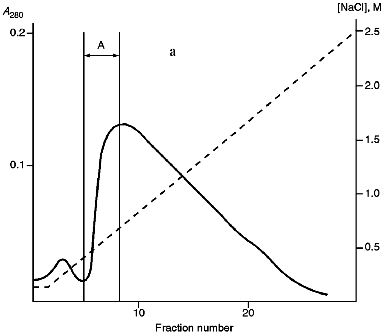
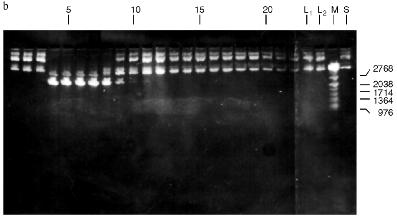
Fig. 4. Purification of endonuclease BspMKI on heparin-Sepharose column: a) optical elution profile (A280) of column. Solid line shows optical density; dashed line indicates NaCl molarity in the gradient fractions. A, fractions containing BspMKI; b) analysis of endonuclease activity in gradient fractions. All other designations are the same as in Fig. 3.
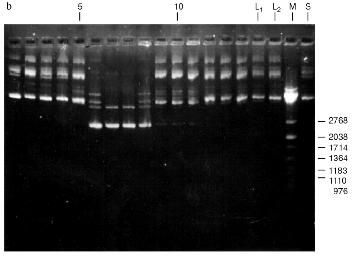
Determination of Substrate Specificity. Substrate specificity of this endonuclease was determined using a set of linear DNA forms of pJRD184 cleaved by unique sites (Fig. 6). Electrophoretic separation of products of pJRD184 hydrolysis cleaved via unique sites and by BspMKI revealed that the site sought for is located about 300 nucleotide pairs from the site BamHI, less than 100 nucleotide pairs from the site SphI, about 900 nucleotide pairs from the site BspLU11II, about 1250 nucleotide pairs from the site XhoI, about 3100 nucleotide pairs from the site EcoRI, and about 800 nucleotide pairs from site PstI. Analysis of cleavage products revealed that BspMKI recognizes the site corresponding to the site of endonuclease SalI (5´-GTCGAC-3´) [5, 6].Fig. 5. Test of restriction--ligation--restriction: 1) plasmid pBR322 DNA (control); 2) pBR322 DNA cleaved by BspMKI; 3) ligated plasmid DNA after cleavage by BspMKI; 4) product of ligation reaction (lane 3); 5) markers of fragment lengths (phage lambda DNA cleaved by BbvII).
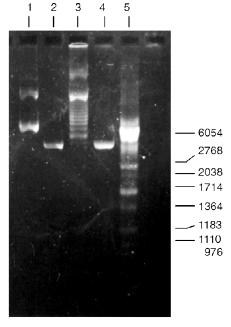
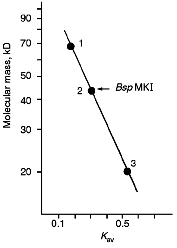
Molecular mass of BspMKI was determined by gel-chromatography on a Sephacryl S200HR column. The enzyme was eluted in a volume that corresponded to a molecular mass of 43-45 kD (Fig. 7).Fig. 6. Cleavage of various plasmid pJRD184 and its linear forms by endonuclease BspMKI. The plasmid was prehydrolyzed by site-specific endonucleases catalyzing a single-point cleavage: 1) BamHI; 2) SphI; 3) BspLU11II; 4) XhoI; 5) EcoRI; 6) PstI and then treated by BspMKI; 7) pJRD184 cleaved by BspMKI. M, markers of fragment lengths (phage lambda DNA cleaved by BbvII); S, substrate DNA pJRD184 without cleavage.
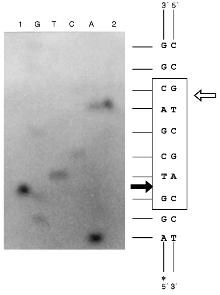
Determination of Cleavage Points on DNA by Endonuclease BspMKI. Phage M13tg130 DNA was used as a matrix for determination of cleavage points. It contains a BspMKI recognition site near the universal primer. Figure 8 shows results of sequencing. Restrictase BspMKI recognizes site 5´-GvTCGAC-3´ and cleaves it inside the recognized sequence after the first significant nucleotide as the arrow indicates. Thus, restrictase BspMKI is an isoschizomer of endonuclease SalI [7].Fig. 7. Determination of molecular mass of BspMKI by eluting volume from a Sephacryl S200HR column. Coefficient Kav was determined as: Kav = (Vr - V0)/(Vt - V0), where Vr is elution volume, V0 is free column volume, and Vt is total volume. Marker protein: 1) albumin (68 kD); 2) ovalbumin (43 kD); 3) trypsin inhibitor (20 kD).
Some Properties of Endonuclease BspMKI. For elucidation of optimal conditions for the restriction reaction we varied ionic strength, pH of the buffer for restriction, and the incubation temperature. NaCl concentrations in the reaction buffer (10 mM Tris-HCl, pH 7.4, 100 µg/ml albumin, 10 mM MgCl2) were 10, 50, and 100 mM for LRB, MRB, and HRB [8], respectively. Values of pH of the reaction buffer were 7.0, 7.4, 8.0. Incubation temperatures were 37, 48, 55, and 65°C. This site-specific endonuclease is characterized by the following parameters: 1) optimal buffer for this site-specific endonuclease is MRB; 2) optimal pH for the enzymes is 7.4; 3) optimal temperature of the incubation is 55°C. The isolated restrictase is a very stable enzyme. Storage at room temperature during two weeks did not influence its activity.Fig. 8. Determination of cleavage points of DNA by endonuclease BspMKI on phage M13tg130 DNA. G, A, T, and C are sequencing lanes; 1) product of polymerase reaction cleaved by BspMKI; 2) the same product after the treatment by Klenow fragment DNA polymerase I in the presence of triphosphates. Frame designates recognition site; dark and light arrows show cleavage point in the sequenced and complementary chains, respectively.
DISCUSSION
The present report describes a scheme of isolation and purification of site-specific endonuclease BspMKI from the thermophilic strain Bacillus species MK. Endonuclease BspMKI was purified to functional homogeneity. Criteria of functional purity are: 1) good ligation of products of substrate DNA hydrolysis after cleavage by BspMKI and formation of homogenous fragments during repeated restriction of ligated products; 2) absence of endonuclease contamination tested during long-term incubation of DNA with excess of the enzyme; 3) absence of 5´-phosphatase and
5´-exonuclease activities in sequencing experiments of recognized primers using 5´-terminus labeled primer. The isolated site-specific endonuclease is an isoschizomer of endonuclease SalI and it recognizes the sequence 5´-GvTCGAC-3´. The molecular mass of endonuclease BspMKI determined by gel-chromatography on Sephacryl S200HR is about 45 kD. The optimal reaction buffer for BspMKI is MRB buffer, pH 7.4, and the optimal incubation temperature is 55°C.
The yield of BspMKI is 70,000 units from 9 g of biomass. Thus, the strain Bacillus species MK is good producer of this endonuclease. The enzyme is also characterized by good stability: two-week storage at room temperature does not decrease its activity. Site-specific endonuclease BspMKI is most active at 55°C. The analog of this enzyme, SalI, is active at 37°C and is inactivated at 65°C, whereas BspMKI still retains some activity at this temperature. Total inactivation of BspMKI occurs at 75°C. These properties suggest BspMKI as a promising enzyme for use in gene-engineering works.
This work was supported by the Russian Academy of Sciences and Russian Foundation for Basic Research (grant No. 96-04-49050).
LITERATURE CITED
1.Roberts, R. J., and Macelis, D. (1992) Nucleic
Acids Res., 20, Suppl., 2167-2180.
2.Brown, N. L., and Smith, M. (1980) Meth.
Enzymol., 65, 391-404.
3.Kieny, M. P., Lathe, R., and Lecocq, J. P. (1983)
Gene, 26, 91-99.
4.Skoups, R. (1985) Methods of Protein
Purification [Russian translation], Mir, Moscow.
5.New England Biolabs Catalog (1996/1997) New
England Biolabs Inc., Beverly.
6.Molecular Biology: Catalog and Product
Application Guide, (1996/1997) MBI Fermentas Inc., Vilnius.
7.Arrand, J. R., Myers, P. A., and Roberts, R. J.
(1978) J. Mol. Biol., 118, 127-135.
8.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor Lab. Press, Cold Spring Harbor.