DNase II in Spermatozoa of the Loach Misgurnus fossilis L.
Yu. V. Nechaevsky1* and V. A. Ivanov2
1Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia; fax: (27) 79-05532Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia; fax: (095) 923-3602; E-mail: ivanov@ibpm.serpukhov.su
* To whom correspondence should be addressed.
Received October 19, 1998; Revision received February 11, 1999
A deoxyribonuclease (DNase) which is active at acid pH in the absence of bivalent cations was found in loach spermatozoa. The enzyme was purified by ion-exchange chromatography and partially characterized. The DNase has optimal activity at pH 5.5 and its molecular weight is about 30 kD; its substrate is covalently closed circular duplex DNA, and its product is the corresponding unit-length linear DNA. The DNase is inhibited by MgCl2 and activated by EDTA. Thus, this endoDNase found in loach spermatozoa can be classified as a DNase II. The biological role of this DNase II is discussed.
KEY WORDS: loach spermatozoa, acid DNase, chromatographic purification, enzyme properties
Endodeoxyribonuclease II (EC 3.1.22.1), which optimally splits intra-chain phosphodiether bonds in duplex DNA at acidic pH in the absence of bivalent cations, has been found in the cells of various organisms [1, 2]. The function of acidic DNase is thought to be the degradation of foreign DNA since the enzyme is localized in lysosomes, which contain all enzymes necessary for degradation of nucleic acids [3, 4]. Current investigations of spontaneous (programmed) cell death have raised the question of a nuclear pool of DNase II which has been found in animal cells [5-9]. It has been shown that nuclear DNase II is involved in apoptosis (the biological mechanism of cell elimination as a response to specific physiological stimulants) in chinese hamster ovary (CHO) cells [10]. This work has stimulated study of the biological role of DNase II in various organisms and cells. The biological material being investigated is a critical factor in the elucidation of enzyme functions. Spermatozoa are characterized by a specific chromatin structure and the lack of many DNA functions; thus, they are a convenient model system for the investigation of functions of enzymes participating in DNA metabolism in non-divisible cells [11]. However, DNase II activity was not detected in bovine and rabbit spermatozoa [12, 13].
We previously detected a seasonal (physiological) relaxation in vivo of supercoiled DNA in spermatozoa of loach (Misgurnus fossilis L.). This process evidently initiates the elimination of spermatozoa which are not involved in fertilization [14]. We described a DNase which could catalyze such relaxation in a preliminary report [15]. In the present work, we describe the isolation, purification, and properties of the enzyme which allows us to identify it as DNase II. On the strength of these data, we suggest that the functions of this DNase II can be the physiological elimination of mature spermatozoa.
MATERIALS AND METHODS
Reagents and measurements. The following reagents were used for enzyme purification and analysis of its activity: DEAE-cellulose and phosphocellulose P11 from Pharmacia (Sweden); polyethylene glycol 600 (PEG) from Loba-Chemie, Wien-Fischamend (Austria); dithiothreitol (DTT) from Koch Light (England); bovine serum albumin (BSA) from British Drug House (England); all other reagents (agarose, Na2EDTA, glycerol, nonionic detergent NP-40, beta-mercaptoethanol (beta-ME), SDS) were from Sigma (USA). Supercoiled pUC19 and pBR322 plasmid DNA prepared by the alkaline method [16] served as the substrate. Protein concentration was measured by the Bradford method [17], and DNA was determined by the diphenylamine reaction [18].
Animals and isolation of spermatozoa. Male loach (Misgurnus fossilis L.) were caught during the autumn and winter period [14] and were continually kept at 2-4°C. Testes were separated, and isolated spermatozoa were washed three times with cold (2°C) solution of 140 mM NaCl using centrifugation (200g, 5 min).
The degree of differentiation and homogeneity of the resulting cell fraction were determined by flow cytofluorometry and scanning electron microscopy. In the first case cells were fixed in 50% ethanol, harvested by centrifugation, and suspended in 100 mM Tris-HCl buffer (pH 7.4) with 100 mM NaCl and fluorescent probe Hoechst-33258 (Calbiochem, USA) at concentration 1 µg/ml. After 15 min of incubation, fluorescence was measured using the original LAKS-1 flow cytofluorometer [19]. The intensity of the stained cell fluorescence was proportional to the DNA content in these cells. The cells were also analyzed using scanning electron microscopy [20] using a JSM-U3 scanning electron microscope (Japan Electron Optics Laboratory, Japan).
Determination of acidic DNase activity. DNase activity was determined by the conversion of native closed circular (supercoiled double-stranded) DNA to its relaxed form or linear DNA of the same size. The reaction mixture contained 0.7 µg of supercoiled plasmid DNA, 10 mM CH3COONa (pH 5.5), 80 mM NaCl, 1 mM Na2EDTA, and BSA (170 µg/ml). The reaction was conducted at 14°C for 15 min in 10 µl of reaction mixture. Products of the reaction were analyzed by electrophoresis in 1% agarose gel. Electrophoresis was carried out in Tris-borate buffer containing 89 mM Tris-borate (pH 8.0) and 2 mM Na2EDTA at 5.2 V/cm for 2.5 h. The amount of enzyme converting 1 µg of supercoiled DNA to the linear form under these conditions was taken as 1 unit of enzyme activity.
Purification of DNase from spermatozoa. All procedures were carried out at 0-4°C. Cells (6·109; ~5.7 mg protein) were lysed in 1 M NaCl in 100 ml of buffer A (10 mM CH3COONa, pH 5.5, 1 mM Na2EDTA, 1 mM DTT) (fraction 1). DNA was removed by precipitation in 6% PEG with subsequent centrifugation according to [21]. The supernatant was dialyzed against buffer B (50 mM Tris-HCl, pH 7.12, 1 mM Na2EDTA, 5 mM beta-ME, 0.1% NP-40, 20 mM NaCl, 5% glycerol) for 15 h. The resulting fraction 2 with low salt concentration was applied to a DEAE-cellulose column (1.4 × 6 cm) equilibrated with buffer B. The column was washed with two volumes of the same buffer. Proteins were eluted with a linear gradient of NaCl (20-900 mM) in 200 ml of the same buffer. Protein concentration and DNase activity were determined in all fractions. Active fractions (eluted at 250-290 mM NaCl) were combined and dialyzed against buffer B with 10% glycerol minus NaCl. The resulting protein solution (fraction 3) was applied to a phosphocellulose P11 column (0.8 × 7 cm) equilibrated with buffer B. The column was washed with two volumes of buffer B and the enzyme was eluted with a linear gradient of NaCl (50-900 mM) in 50 ml of the same buffer. Fractions (0.5 ml) which were eluted by 500-540 mM NaCl were combined and dialyzed against 40% glycerol in 50 mM Tris-HCl (pH 8.0) with 0.2% NP-40. The purified enzyme (fraction 4) was stored at -20°C.
Enzymatic characteristics of the DNase. The effects of ionic strength and metal cations on the spermatozoon DNase activity as well as the molecular weight and optimal pH for activity of the enzyme were studied according to [22-25].
Other methods. Cell lysates were analyzed under standard conditions for DNA-topoisomerases I and II as described in [21, 26].
RESULTS
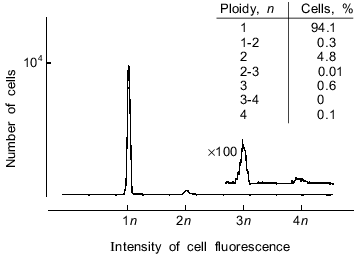
Analysis of the cell fraction. Cytofluorometric analysis of the data showed that at least 94% of the studied cell fraction are haploid cells (Fig. 1). It should be note that in various subfractions of the remaining 6% of cells containing 2 n (or more) DNA some or all of the apparently polyploid cells are actually spermatozoa which have aggregated during sample preparation and/or during measurements.
Morphological features of all viewed cells (reduced cytoplasm which "flows" from head to tail, highly condensed chromatin, a completely formed tail) show that the cell fraction was homogeneous and contained mature spermatozoa (data not shown).Fig. 1. Distribution of cells from the studied spermatozoon fraction by DNA content expressed in terms of ploidy. The original LAKS-1 flow cytofluorometer was used; lambdaex = 365 nm, lambdaem = 460 nm (the procedure is described in detail in "Materials and Methods" and in [19]). Loach erythrocytes with 2 n DNA were used as a control for DNA content in cells. ×100, this part of the histogram was measured with 100-fold increase in sensitivity; 104 cells were analyzed.
Identification of DNase activity. We previously detected a seasonal (physiological) in vivo relaxation of supercoiled DNA in loach spermatozoa. This process seems to be the part of the elimination of the spermatozoa which do not participate in fertilization [14]. Here we have tried to identify enzymatic the activity responsible for this relaxation. To do this, cell lysate was analyzed under standard conditions optimal for exhibition of DNA-topoisomerase I and II activities as well as for activities of Mg2+/Ca2+- (or Mg2+)-dependent (and independent) endonucleases at acidic and alkaline pH values. Only DNase activity which is exhibited at acidic pH values and in the absence of bivalent cations was found. Alkaline (Mg2+- and/or Ca2+-dependent) DNase and DNA-topoisomerase activities were not found. Previously, DNase II-like DNase activity was found in the sperm of two mammalian species, bull and rabbit [12, 13], and in testes of fishes, mackerel and salmon [2, 27], but it had not been detected in spermatozoa. Thus, we decided to try to isolate and characterize DNase from loach spermatozoa.
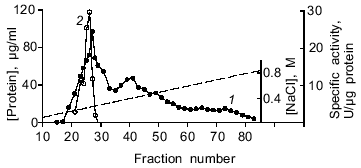
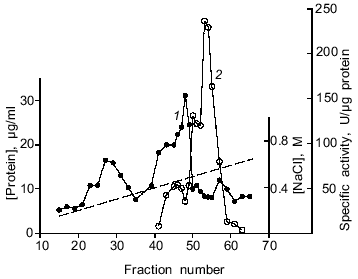
Purification of DNase II. The results of a typical spermatozoon DNase purification are given in the table, and the procedure was described in detail in "Materials and Methods". The purification process includes chromatography on DEAE-cellulose and phosphocellulose. Such ion-exchange chromatography was previously used for isolation of acidic DNases from various tissues of mammals, birds, and fishes [2, 28, 29]. The enzyme of spermatozoa was eluted from a DEAE-cellulose column (Fig. 2) and a phosphocellulose column (Fig. 3) with 270 and 520 mM NaCl, respectively. However, a DNase II from porcine spleen eluted in 250 mM phosphate buffer [28] and a human enzyme from mucous ventricle membrane or uterus cervix existing in two forms was eluted from phosphocellulose in 350 (minor peak of activity) and 400 mM (the main peak of activity) KCl [29]. Thus, the loach enzyme distinctly differs from the human acidic DNase in its behavior during chromatography on phosphocellulose. After ion-exchange chromatography, the overall purification of the enzyme was 139-fold (in relation to the initial cell lysate) with 22% yield (table). The molecular weight of the only polypeptide component of the enzyme preparation (fraction 4 after chromatography on phosphocellulose) was determined using SDS-PAGE in a gradient gel. It was ~30 kD (data not shown). This value is comparable with data obtained for DNase II from the other organisms. For example, native bovine enzyme (40-45 kD) comprises two subunits (30-35 and 10 kD); a lesser band colors weakly and can be hardly noticed in SDS-PAGE [30, 10]. The value obtained is consistent with the molecular weight of DNase II from CHO cells, which participates in apoptosis of these cells [10].
Fig. 2. Chromatography of dialyzed loach spermatozoon proteins on DEAE-cellulose (nuclease activity was measured at pH 5.5 in the absence of metal ions with supercoiled plasmid DNA as the substrate): 1) protein concentration in fractions; 2) specific DNase activity in µg supercoiled DNA linearized per µg fraction protein (see "Materials and Methods").
Purification of DNase from loach spermatozoaFig. 3. Chromatography of a partially purified preparation of acidic DNase from loach spermatozoa on phosphocellulose P11. Active fractions eluted from DEAE-cellulose at 250-290 mM NaCl were used. All designations as in Fig. 2.
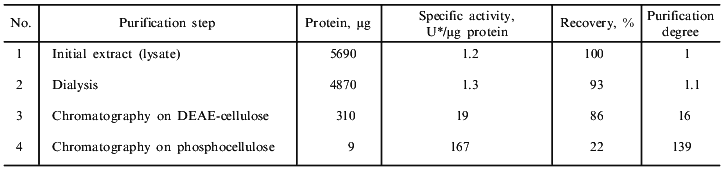
*The amount of enzyme converting 1 µg of supercoiled plasmid DNA to its linear form at 14°C in 15 min was taken as 1 unit of enzyme activity.
Properties of the purified enzyme. The nucleolytic activity with closed circular double-stranded DNA as the substrate shows that the enzyme is an endonuclease. The spermatozoon DNase shows linear kinetics of the nuclease reaction in the range from 0 to 15 min in a standard reaction mixture (Fig. 4). The product of the enzymatic reaction under the described conditions is linear DNA. Traces of opened circular DNA with a nick can be detected only after prolonged incubation (60 min or more, Fig. 4). The formation of these molecules can be explained by spontaneous hydrolysis, which can take place under these conditions; specifically, acidic pH value causes loss of DNA bases [31]. These data are consistent with a known property of DNase II: the enzyme degrades DNA mainly by the diplotene mechanism [3]. Hence, the enzyme mainly attacks double-stranded DNA. The activities of some DNases II (including the enzyme from salmon testes) toward native (double-stranded) and thermally denatured (single-stranded) DNA differ 10-15-fold [27, 29].
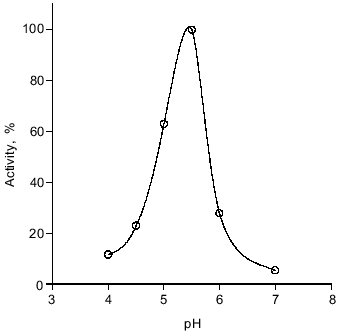
The enzyme was active in the temperature range from 0 to 40°C with maximal activity at 14°C. It is notable that at 0°C its activity was no less than 20% of the optimal activity (data not shown). The DNase from spermatozoa exhibits maximal activity at pH 5.5. At pH 6 and 4 the rate of hydrolysis is 27 and 11% of the maximal rate, respectively (Fig. 5). The enzyme exhibits maximal activity in the absence of bivalent cations: 5 mM Mg2+ inhibits the nuclease reaction fourfold (Fig. 6). Consistent with this observation, low concentrations of Na2EDTA weakly stimulate the reaction, presumably binding endogenous bivalent cations.Fig. 4. Kinetics of the nuclease reaction catalyzed by the DNase from loach spermatozoa. Enzymatic activity was as the conversion of native closed circular supercoiled DNA to its linear form (1) or opened circular (relaxed) DNA (2). The activity of the enzyme after 2 h of conversion was taken as 100%.
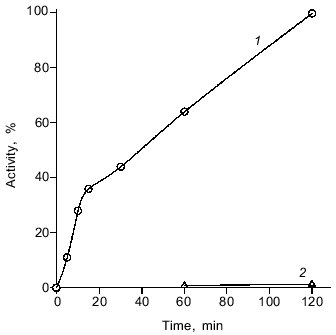
Fig. 5. Dependence of DNase activity on pH. Loach spermatozoon DNase activity was determined in the absence of metal ions at the indicated pH values. A standard reaction mixture was used (see "Materials and Methods"). The activity at the pHopt was taken as 100%.
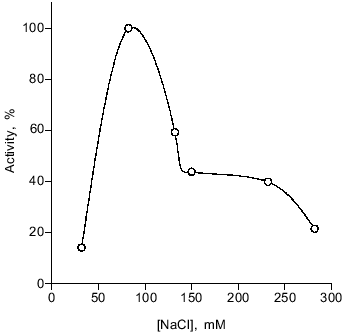
The optimal NaCl concentration for activity is 80 mM. The enzyme is not very sensitive to high ionic strength, retaining 40-45% of its maximal activity in 150-230 mM NaCl (Fig. 7), whereas the salmon enzyme was totally inhibited in 250 mM CH3COONa [27].Fig. 6. Influence of Mg2+ on activity of the loach spermatozoon DNase. The enzyme activity under standard conditions (in the absence of Mg2+) was taken as 100%.
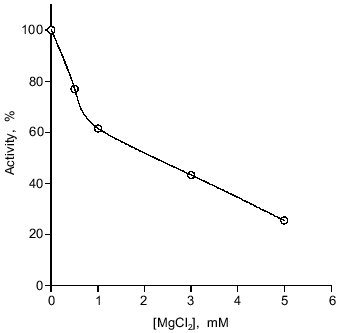
Fig. 7. Influence of ionic strength on activity of loach spermatozoon DNase. The activity of the enzyme under standard conditions (at 80 mM NaCl) was taken as 100%.
DISCUSSION
We have studied some properties of a DNase from loach spermatozoa. The enzyme hydrolyses supercoiled closed circular DNA at acidic pH values in the absence of bivalent cations; its molecular weight is ~30 kD. Mg2+ inhibits the enzymatic activity. On the basis of the studied properties, the isolated DNase of spermatozoa is a DNase II-type enzyme; such enzymes are widespread in cells of various organisms [1, 2]. This DNase was also found in animal testes, including mackerel [2] and salmon [27], and in bovine [17] and rabbit [13] sperm, but it was not found in spermatozoa of mammals [12, 13]. While looking for enzymatic activities relaxing DNA of loach spermatozoa in vivo, we detected the only activity corresponding to the DNase II. Thus, it appears that the DNase II is the only enzyme catalyzing the breakdown of nucleic acids in the spermatozoa of these fish.
DNase II was previously believed to be lysosomal enzyme. Lysosomes contain a set of enzymes required for degradation of nucleic acids to nuleosides and, therefore, it was considered that DNase II plays a degradative (digestive) role characteristic of lysosomal enzymes secreted by pancreatic cells [3]. However, the DNase II was detected in the nucleus; this suggests that this enzyme has another biological function [5-9]. These investigations demonstrate an apparent connection between the level of a nuclear pool of DNase II and synthesis of DNA during organogenesis or tissue regeneration.
Another (non-digestive) function of DNase II can be inferred from investigation of this enzyme from testes and spermatozoa of animals. Acidic DNase was found in mammalian sperm 35 years ago [12]. The reports of this enzyme from mackerel testes [2] and human sperm [32] appeared later. In a recent study of DNases from reproductive organs of male rabbits, it was demonstrated that the activity of DNase II was 2-5-fold higher in testes, epididymis, prostate, and sperm than in kidney, liver, and spleen [13]. Thus, this enzyme may participate in the maturation of spermatozoa. Finally, in the present work a DNase II from loach spermatozoa has been described as the only enzyme catalyzing the breakdown of nucleic acids in these highly specialized, terminally differentiated cells.
It was recently demonstrated that DNase II takes part in apoptosis--the programmed cell death which can be stimulated by specific physiological stimulants or cytotoxic agents [10]. It seems likely that in some cell types DNase II is the main nuclease participating in the mechanism of apoptosis (it causes inter-nucleosomic digestion of DNA). It seems that this biological function of the enzyme adequately explains the nuclear localization (the nuclear pool) of DNase II as well as the correlation between the activity of nuclear DNase II and DNA synthesis (and high activity of this enzyme in reproductive organs). It is known that the physiological death of differentiating spermatogonia in normal testes is due to apoptosis [33]. Because of this, it can be assumed that biological function of DNase II in mammalian reproductive organs consists in provision of negative selection of spermatogonia during spermatogenesis or in activation (elimination) of epididymal maturation. The results of the present study--the identification of a DNase II in loach spermatozoa--suggest the possibility that negative selection of spermatozoa in fish may occur directly at the level of the mature spermatozoa.
REFERENCES
1.Allfrey, V., and Mirsky, A. E. (1952) J. Gen.
Physiol., 36, 227-241.
2.Gordonnier, C., and Bernardi, G. (1968) Can. J.
Biochem., 46, 989-995.
3.Bernardi, G. (1971) The Enzymes, 4,
271-287.
4.Liao, T.-H., Liao, W.-C., Chang, H.-C., and Lu,
K.-S. (1989) Biochim. Biophys. Acta, 1007, 15-22.
5.Swingle, K. F., and Cole, L. J. (1964) J.
Histochem. Cytochem., 12, 442-447.
6.Lesca, P. (1968) Nature, 220,
76-77.
7.Slor, H., and Lev, T. (1971) Biochem. J.,
123, 993-995.
8.Slor, H. (1973) Biochem. J., 131,
83-87.
9.Slor, H., Bustan, H., and Lev, T. (1973)
Biochem. Biophys. Res. Commun., 52, 556-561.
10.Barry, M. A., and Eastmann, A. (1993) Arch.
Biochem. Biophys., 300, 440-450.
11.Grimes, S. R. (1986) Comp. Biochem.
Physiol., 83, 495-500.
12.Waldschmidt, M., Karg, H., and Kinzler, M. (1964)
Naturwissenschaft, 51, 364-365.
13.Takenshita, H., Yasuda, T., Nadano, D., Tenjo,
E., Sawazaki, K., Iida, R., and Kishi, K. (1994) Int. J.
Biochem., 26, 1025-1031.
14.Nechaevsky, Yu. V., Strazhevskaya, N. B., and
Kuzin, A. M. (1983) Stud. Biophys., 93, 9-16.
15.Nechaevsky, Yu. V., and Ivanov, V. A. (1995)
Dokl. RAN, 343, 551-554.
16.Maniatis, T., Fritsch, E. F., and Sambrook, J.
(1982) Molecular Cloning, Cold Spring Harbor Laboratory, pp.
90-94.
17.Bradford, M. M. (1976) Anal. Biochem.,
72, 248-254.
18.Croft, D. N., and Lubraw, M. (1965) Biochem.
J., 95, 612-620.
19.Pechatnikov, V. A., Afanasyev, V. N., Korol, B.
A., Korneev, V. N., Rochev, Yu. A., and Umansky, S. R. (1986) Gen.
Physiol. Biophys., 5, 273-284.
20.Goldstein, S. I., Newbury, D. E., Ehlin, P., Soy,
D. C., Fiovi, C., and Lifchin, E. (1981) Scanning Electron Miroscopy
and X-Ray Microanalysis, Plenum Press, N. Y.
21.Ivanov, V. A., and Melnikov, A. A. (1984) FEBS
Lett., 177, 300-304.
22.Ivanov, V. A., Gaziev, A. I., and Tretyak, T. M.
(1983) Eur. J. Biochem., 137, 517-522.
23.Ivanov, V. A., Tretyak, T. M., Terpilovskaya, O.
N., and Smirnova, G. N. (1987) Biokhimiya, 52,
842-845.
24.Ivanov, V. A. (1987) Biokhimiya,
52, 1133-1137.
25.Ivanov, V. A., Tretyak, T. M., and Afonin, Yu. N.
(1988) Eur. J. Biochem., 172, 155-159.
26.Ivanov, V. A., Melnikov, A. A., and Terpilovska,
O. N. (1986) Biochim. Biophys. Acta, 866, 154-160.
27.Yamamoto, M. (1971) Biochim. Biophys.
Acta, 228, 95-104.
28.Bernardi, G., Bernardi, A., and Chersi, A. (1966)
Biochim. Biophys. Acta, 129, 1-11.
29.Yamanaka, M., Tsubota, Y., Anai, M., Ishimatsu,
K., Okumura, M., Katsuki, S., and Takagi, Y. (1974) J. Biol.
Chem., 249, 3884-3889.
30.Liao, T.-H. (1985) J. Biol. Chem.,
260, 10708-10713.
31.Lindahl, T., and Nyberg, B. (1972)
Biochemistry, 11, 3610-3618.
32.Quinn, P. J. (1968) J. Reprod. Fertil.,
17, 35-39.
33.Allan, D. J., Harmon, B. V., and Kerr, J. F. R.
(1987) in Perspectives on Mammalian Cell Death (Potten, C. S.,
ed.) Oxford University Press, N. Y., pp. 229-258.