A Chapter from the Book of V. N. Manskikh “Essays of Evolutionary Oncology” (Perelmuter, V. M., ed.) published in 2004 by Siberian Medical University Publisher, Tomsk
2.8. Tumor Growth as a Regulatory Mechanism of Mutational Burden in Populations
V. N. Manskikh
DOI: 10.1134/S000629790608189
The emergence and development of a blastoma, whatever its origin, are underlain by mutational changes in the cell genome leading to activation of protooncogenes. From the viewpoint of population genetics, oncogenesis can be considered as a lethal somatic mutation. However, these mutational changes are insufficient for development of a tumor. A number of anti-oncogenic systems are to be deactivated, first of all the recently discovered system of apoptotic cell death (N. V. Berezhkov, 1990).
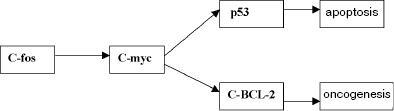
The molecular system triggering apoptosis is known to provide for the stability of the somatic cell genome of the tissue of organisms due to activation of mechanisms of programmed death of cellular elements in the case of a considerable number of mutation-induced damages (E. B. Vladimirskaya et al., 1997). The apoptosis system is functionally associated with the oncogenesis system and also with the systems of DNA repair, because it controls the efficiency of the latter (I. I. Belushkina, S. E. Severin, 2001). Apoptotic death and malignization are only two possible final results of the same cascade of genetic changes (Fig. 2).
It should be noted that apoptosis is triggered if the system of genetic damage repair is damaged (K. Yu. Luk'yanova et al., 2000), and in this case one type of repair (post-replication repair) is realized when two other types (excision repair and photoreactivation) are insufficient, i.e., this system has a definite functional hierarchy. Moreover, mutant cells are eliminated by various immune mechanisms and other anti-mutagenic systems (responsible for elimination and recovery which overall have a hierarchic organization and act on particular organic levels (N. I. Goncharova, 1984; N. N. Il'inskikh et al., 1992)). The functioning of all these systems results in the regulation of mutation frequencies in the somatic and generative cells and, consequently, the mutational burden in the population. To demonstrate the functioning of this system and the place of tumor growth in it, consider the situation of a dramatic increase in the frequency of “spontaneous” mutations induced in natural populations by an increased mutagenic effect of environmental factors. These mutations can include a noticeable number of lethal and sublethal alleles which, on flowing into the population, can decompensate the genetic stability and result in a dramatic decrease in the numbers of a species and its biological regression (A. A. Korol'kov, V. P. Petlenko, 1977; A. V. Yablokov, A. G. Yusufov, 1981)1.Fig. 2. Simplified scheme of molecular mechanisms of activation of apoptotic processes and malignant degeneration of the cell (E. A. Kogan, 1996).
1 In this connection, experiments are demonstrative of the well-known Russian geneticist A. S. Serebrovsky on the successful use of lethal mutations to decrease the numbers of harmful insect populations. It is known that unrestricted variability can very easily destroy the most sophisticated interactions inside organic systems, from interactions of molecules inside cells to those of individuals in populations and populations in biocenoses (A. I. Strukov et al., 1983). However, this usually does not occur, because various mechanisms exist which provide for the stability of the population's genetic structure.
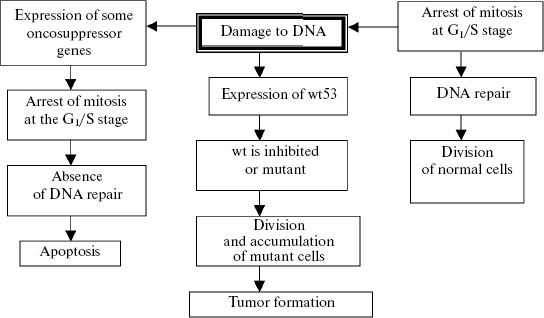
If the number of damages is not very significant, they are eliminated by excision or photorepair, or compensated by conversion to the heterozygous state and introduced into the general genofond of a population as a source of genetic variability and material for natural selection (on the somatic cell level this process is expressed by polyploidization (or amplification) with subsequent diminution of the defective chromatin). Mutations not eliminated by these mechanisms undergo the post-replication repair, or the mutant cells are removed by mechanisms unassociated with mutagenesis (differentiation of cells with the defective genome (J. E. Evans, 1976, cited after N. N. Il'inskikh et al., 1992)). If the abovementioned systems are functionally incompetent, a chain of genetic changes is triggered which result in the apoptotic death of the cells with the excess mutational burden. Special mechanisms are known which control the efficiency of DNA repair. If the repair mechanisms are insufficient during genome replication, an enzymatic cascade is switched on after the cell cycle has been arrested at the G1/S stage and unrepaired damages of the genome have been detected, and this results in internucleosomal degradation of chromatin, i.e., apoptosis (K. Yu. Luk'yanova et al., 2000). If this mechanism is also damaged (for instance, the gene p53 mutates to the gene wt53), the cascade will be terminated not by apoptosis but by activation of protooncogenes and transformation of a normal cell into a malignant one (Fig. 3).
Tumor progress will result in the death of an individual whose genome can be dangerous for the stability of the population's genofond because of serious damage. Certainly, this mechanism is nonspecific, because it reacts not to the quality of mutation-induced changes (but only those which do not affect the “apoptosis-oncogenesis” system links because they belong to definite genes, and in this respect oncogenesis is specific), but to the strength of the mutational effect of a carcinogenic agent. This mechanism can be roughly likened to the fuse filament, which is burnt away because of high voltage and disables the fuse and, thus, saves the electric apparatus. The oncogenesis systems are triggered probabilistically: an increase in the mutation-inducing effect is accompanied by an increase in the probability of damaging protooncogenes and genes triggering the apoptosis system (first of all, p53) and, consequently, also in the probability of mutations in the generative cells. Due to this probabilistic dependence, the mutation frequency in the generative cells of individuals in the certain population is corrected by mutations in the somatic cells (protooncogenes). This effect is very illustratively exemplified by retinoblastomas emerging because of loss of gene RB (including the generative cells) and also by tumors in persons with the affected mechanisms of DNA repair, e.g., with xeroderma pigmentosum or chromosome instability. The increase in the frequency of tumor neoplasms with age is a similar phenomenon, and this frequency is rather high in the age when the reproductive activity is still fully retained, although its peak falls on old age in connection with accumulation of many mutational changes in the genome. Data on the stepwise molecular changes during carcinogenesis also indicate an important role of tumor growth in the regulation of the mutagenesis intensity. The number of mutations resulting in tumor development has to be no less than two or three, but more often they are five or six (P. D. Lawley, 1994). These are mutations of protooncogenes, genes of tumor suppression, and mismatch-repair genes (W. K. Cavanee, R. L. White, 1995). Studies on the ratio between the DNA repair levels and natural (“background”) mutagenesis have revealed that the latter cannot supply more than two or three successive mutations (L. A. Loeb, 1991; D. N. Cooper, M. Krawczak, 1993). Thus, spontaneous mutagenesis cannot provide the sufficient number of consecutive mutations required for carcinogenesis. The role of such inducers which, under certain conditions, increase the frequency of mutations is played by special mutators. It is just their activation that is the mechanism responsible for reaching the necessary level of genetic instability of the gene cascade, the damage of which leads to development of tumor (L. A. Loeb, 1991). Hence, variability of the genes influencing blastomogenesis is activated by the state of the cell genome; therefore, damage to these genes cannot be considered only as a spontaneous and independent process.Fig. 3. Scheme of oncosuppressor gene wt53 functioning in the case of DNA damage or on receiving a specific signal (N. T. Raikhlin, A. N. Raikhlin, 2002).
Naturally, anti-mutagenic mechanisms in multicellular animals are not limited only to apoptosis. The number of cells with the affected system of apoptosis and activated system of oncogenesis, which are potentially malignant and constantly produced in humans, is rather significant (J. Folkman, 1974), but tumors are a relatively rare phenomenon. This is due to the mechanisms of nonspecific immunity, which eliminates the arising mutant cells (macrophages, NK cells). Depression of the immune system (in different immunodeficiencies) results in a dramatic increase in the number of neoplasms, i.e., throwing out individuals with immunity defects through tumorigenesis. Thus, there is a certain hierarchy of eliminating anti-mutagenic mechanisms which are causally interrelated and responsible for functioning on the definite level of organization: 1) the molecular level, or DNA repair (A is excision repair and photoreactivation, B is a post-replication repair); 2) the cellular level, or “endogenous” apoptosis; 3) the tissue level, or the cell elimination through immunocyte action; 4) the organism's level, or the individual's death caused by tumor progress; 5) the population's level, or factors of natural phenotypic selection (Table 1).
Table 1. Structure of the hierarchical
anti-mutagenic system (partially after N. N. Il'inskikh et al.,
1992)
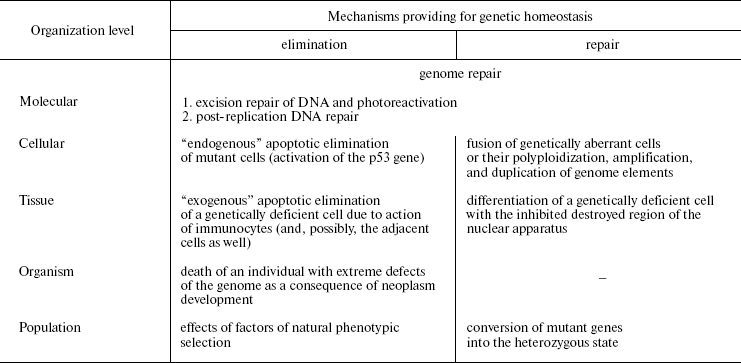
From the foregoing text, it follows that oncogenesis is causally related not only with mutagenesis, but also with the functioning of anti-mutagenic systems that results in restricting the role of blastomas to the regulation of the mutation frequency in populations. Owing to the lethal effect, the tumor progress removes from the population the individuals who a priori possess more severe damage to the reproductive cell genome. From this standpoint, oncogenesis is only an eliminating link in the hierarchical system responsible for regulation of the mutational burden, which acts on the organism's level. The hierarchical character of this system should be once more emphasized: the insufficient functioning of any of its link compensates activities of the other levels. The existence of such a system provides for the regulation of mutation frequency in living beings on different organizational levels, also including populations, and, consequently, mechanisms of evolution.
From the standpoint of this concept, the nonuniform distribution of tumors in phylogenetic series is explained by the different contribution of oncogenesis to the regulation of the genetic stability of a population: in the individuals with a high frequency of spontaneous blastomas, blastomogenesis plays a significant role in these mechanisms.
This concept can be applied not only to chemical and radiation-induced carcinogenesis, but also to viral carcinogenesis, because viruses are mutagenic agents capable of changing the population's genofond (see above notes about oncogenic viruses). The most frequent emergence of tumors in old age does not deprive blastomogenesis of adaptive significance. Obviously, the mutational burden in young animals is markedly lower than in old ones; therefore, extreme mechanisms of its regulation are switched on considerably less often. An increased mutagenic exposure is known to induce a significantly sharper increase in blastoma frequency in young members of the population, because just they are most sensitive to oncogenic influences, and tumors in them are more malignant (L. M. Shabad, 1970). Thus, this primarily removes the objection that the tumor progress can eliminate only old members of the population, who are unable for reproduction. This is especially demonstrative in invertebrates in which the elimination of mutant genotypes by oncogenesis is doubtless: such are Morgan-Stark 1 tumors developing in drosophila larvae and killing male carriers of the mutant genes before their turning to imago, and also similar but less studied tumors in the lepidopterous Pigera nigra (N. N. Petrov, 1947). Due to some specific features of the development cycle of these organisms, the abovementioned adaptive function of tumor growth is displayed in them more distinctly. Just this function connects the tumor growth and fundamental properties of living matter, such as inheritance and variability, the interrelation of which was supposed even by N. N. Petrov (1961). This concept is also closely associated with the recent hypothesis about phenoptosis, or the programmed death of the whole organism (V. P. Skulachev, 1997, 1999), because oncogenesis, similarly to “natural death”, is a mechanism of elimination on the organism's level.
To summarize, it can be concluded that tumor emergence (conversion of protooncogenes into oncogenes) is a mechanism of the hierarchical system regulating the mutational burden in populations, which originated from the ancient excesses of growth and asexual reproduction, and functions on the organism's level. And I think that just this is the evolutionary significance of oncogenesis and, consequently, the conversion of protooncogene to oncogenes.
REFERENCES
1.Berezhkov, N. V. (1990) Arkh. Anat. Gistol.
Embriol., 99, 68-77.
2.Vladimirskaya, E. B., Maschan, A. A., and
Rumyantsev, A. G. (1997) Gematol. Transfuziol., 42,
4-9.
3.Belushkina, I. I., and Severin, S. E. (2001)
Arkh. Patol., 63, 51-60.
4.Kogan, E. A. (1996) in Lectures on
General Pathologic Anatomy (Serov, V. V., and Pal'tsev, M. A.,
eds.) [in Russian], Meditsina, Moscow, pp. 78-90.
5.Luk'yanova, K. Yu., Kulik, G. I., and Chekhun, V.
F. (2000) Vopr. Onkol., 46, 121-128.
6.Goncharova, N. I. (1984) Abst. Papers 14th Conf.
Eur. Soc. Environ. Mutagens, Moscow, p. 199.
7.Il'inskikh, N. N., Novitskii, V. V., Vanchugova, N.
N., and Il'inskikh, I. N. (1992) Micronuclear Analysis and
Cytogenetic Instability [in Russian], Tomsk State University
Publishers, Tomsk.
8.Korol'kov, A. A., and Petlenko, V. P. (1977)
Philosophic Problems of the Theory of Norm in Biology and Medicine
[in Russian], Meditsina, Moscow.
9.Yablokov, A. V., and Yusufov, A. G. (1981) On
Evolutionary Theory [in Russian], Vysshaya Shkola, Moscow.
10.Strukov, A. I., Khmel'nitskii, O. K., and
Petlenko, V. P. (1983) Morphologic Equivalent of Function
(Methodical Principles) [in Russian], Meditsina, Moscow.
11.Evans, J. E. (1976) Cytobiology,
16, 115-124.
12.Raikhlin, N. T., and Raikhlin, A. N. (2002)
Vopr. Onkol., 48, 159-171.
13.Lawley, P. D. (1994) Adv. Cancer Res.,
65, 17-111.
14.Cavanee, W. K., and White, R. L. (1995) Sci.
Am., 3, 50-57.
15.Loeb, L. A. (1991) Cancer Res.,
51, 3075-3079.
16.Cooper, D. N., and Krawczak, M. (1993) Human
Gene Mutation, Bios Scientific Publishers, Oxford.
17.Folkman, J. (1974) Adv. Cancer Res.,
19, 331-358.
18.Shabad, L. M. (1970) Approaches for
Determination and Study of Blastomogenicity of Chemical Compounds
[in Russian], Meditsina, Moscow.
19.Petrov, N. N. (1947) in Malignant Tumors
[in Russian], Vol. 1, Pt. 1, Medgiz, Leningrad, pp. 8-37.
20.Petrov, N. N. (1961) Guidebook on General
Oncology [in Russian], Medgiz, Leningrad.
21.Skulachev, V. P. (1997) Biochemistry
(Moscow), 62, 1191-1195.
22.Skulachev, V. P. (1999) Biochemistry
(Moscow), 64, 1418-1426.