Modulation of Hippocampal Neuron Survival by Thrombin and Factor Xa
L. R. Gorbacheva1, T. P. Storozhevykh2, V. G. Pinelis2, S. Ishiwata3, and S. M. Strukova1*
1Department of Human and Animal Physiology, Biological Faculty, Lomonosov Moscow State University, 119899 Moscow, Russia; E-mail: strukova@mail.ru2Scientific Center for Children's Health, Russian Academy of Medical Sciences, 119991 Moscow, Russia
3Department of Physics, Faculty of Science and Engineering, Advanced Research Institute for Science and Engineering, Waseda University, Tokyo, Japan
* To whom correspondence should be addressed.
Received April 28, 2006; Revision received June 8, 2006
Effects of thrombin, factor Xa (FXa), and protease-activated receptor 1 and 2 agonist peptides (PAR1-AP and PAR2-AP) on survival and intracellular Ca2+ homeostasis in hippocampal neuron cultures treated with cytotoxic doses of glutamate were investigated. It is shown that at low concentrations (<=10 nM) thrombin and FXa protect neurons from glutamate-induced excitotoxicity. Inactivation of the proteases blocked the neuroprotective effect. Using PAR1-AP, PAR2-AP, and PAR1 antagonist, we have demonstrated that the neuroprotective effect of thrombin is mediated through activation of PAR1, whereas the effect of FXa may involve novel subtype(s) of PARs. Unlike FXa, thrombin induced transient intracellular calcium signal in hippocampal neurons, which was mainly mediated via IP3 receptors of the endoplasmic reticulum. Both of the serine proteases improved the recovery of neuronal Ca2+ homeostasis after glutamate treatment.
KEY WORDS: thrombin, factor Xa, glutamate toxicity, apoptosis, intracellular calcium, hippocampal neuronsDOI: 10.1134/S000629790610004X
Abbreviations: ABE1) anion-binding exosite 1; 2-APB) 2-aminoethoxydiphenyl borate; AraC) cytosine arabinoside; FXa) activated coagulation factor X; LDH) lactate dehydrogenase; PAR) protease-activated receptor; PAR1-AP) protease-activated receptor 1 agonist peptide; PMSF) phenylmethylsulfonyl fluoride.
Thrombin (EC 3.4.21.5), a highly specific protease from the trypsin
family and a key protein in the blood coagulation system, is formed as
a result of limited proteolysis of prothrombin by activated factor Xa
at sites of tissue and vascular injury. Thrombin is involved in the
regulation of many physiological and pathophysiological processes by
interactions with protease-activated receptors (PAR1, PAR3, and PAR4)
[1-3]. Thrombin activates
positive and negative feedback reactions in hemostasis and
fibrinolysis, participates in the regulation of vascular tonus,
organism development, as well as in the processes of inflammation,
tissue repair, atherogenesis, carcinogenesis, and neurodegenerative
diseases (Alzheimer's disease and others) [2-6]. Thrombin multifunctionality is associated with the
features of its tertiary structure and the presence of two subsites
(besides the classical active site), so-called anion-binding exosites
(ABE) (also called additional sites for substrate and receptor
recognition) [6, 7].
Anion-binding exosite 2 (ABE2) enables thrombin binding to heparin and
glycosaminoglycans [8]. ABE1, the recognition site
for complementary domains of receptors (PAR1, thrombomodulin), specific
substrates (fibrinogen), and inhibitors is responsible for high
specificity of bond cleavage and selection of thrombin substrates, as
well as thrombin regulatory functions during inflammation and tissue
repair, which are always accompanied by activation of the coagulation
system and thrombin accumulation at the site of injury of vessel and
surrounding tissue [4, 7, 9, 10]. Contemporary data on the
role of thrombin in traumatic and ischemic brain injury are
controversial. Thrombin, whose effect is implicated through the seven
transmembrane G-protein coupled protease-activated receptors (PAR), is
considered both as a death factor and survival factor for neural cells
[10-17]. It has been
demonstrated that the introduction of thrombin in the caudate nucleus
induces the development of inflammation and edema, whereas low enzyme
concentrations have a protective effect on central nervous system cells
[13, 16]. Transient ischemic
episodes in the nervous system result in development of resistance to
subsequent severe brain ischemia, and thrombin is apparently involved
in this phenomenon [16]. Functions of coagulation
factor Xa (FXa), a serine protease that is able to activate PAR1 but
predominantly activates PAR2 [3, 6], are poorly known beyond hemostasis and
inflammation. Expression of prothrombin, FXa, and PAR mRNA was detected
in brain regions most susceptible to ischemic damage, among those being
cortex, striatum, hypothalamus, hippocampus, and cerebellum [13, 17-19].
Data on the role of PAR in regulation of cell survival are also
contradictory. Neurodegenerative function of PAR2 has been reported [20]; however, cell death is diminished upon
activation of these receptors in the course of neuroinflammation,
injury, and ischemia [21-24].
A number of authors indicate increased brain damage upon activation of
PAR1 under conditions of ischemia and hypoxia [24]. It is known that such damages of brain tissue as
trauma, ischemia, and neurodegenerative diseases (Alzheimer's disease
and others) are associated with hyperstimulation of glutamate receptors
[25]. Massive Ca2+ influx into nerve
cells through ionotropic glutamate receptor-associated channels
disrupts the homeostasis of this intercellular messenger, which in turn
results in initiation of a complex apoptotic cell death cascade [26, 27]. Therefore, the study of
reactions triggered in nerve cells by serine proteases during glutamate
toxicity is of special interest.
The aim of this work was to study the effect of thrombin and factor Xa on survival and calcium homeostasis of cultured rat hippocampal neuron cells during glutamate toxicity.
MATERIALS AND METHODS
Hippocampal neuron culture. Studies were performed using 9-14-day primary culture of neurons from hippocampus obtained from brain of 1-3-day-old Wistar rats. Cell suspension was obtained according to a previously described technique [28] and transferred onto a cover glass (one hippocampus in 200 µl per glass) covered with poly-D-lysine (10 mg/ml). The cells were allowed to sediment for 1 h at 37°C and 5% CO2, then non-attached cells were removed and 1.5 ml of culture medium (neurobasal medium A containing 2% Supplement B-27 and 0.5 mM L-glutamine) was added. At day 3-4 cytosine arabinoside (AraC, 10-5 M) was added to suppress the growth of glial cells.
Determination of neuron death. Neuron death in culture was estimated 24 h after the cell exposure (for 15 min) to glutamate and other studied substances, which were added to cells upon replacement of culture medium with HEPES-saline buffer (HBSS). HBSS composition in mM was: NaCl, 145; KCl, 5; CaCl2, 1.8; MgCl2, 1.0; HEPES, 20; glucose, 5 (pH 7.4). Cell death was determined using two approaches, biochemical and morphological. The biochemical approach was based on the measurement of lactate dehydrogenase level (LDH) in the medium. The presence of cytoplasmic enzyme in the culture medium indicates the damage of cell membrane (necrosis). LDH activity was determined spectrophotometrically at 340 nm using an Anthos Lucy1 microplate reader (Anthos Labtec Instruments, Austria) and LDH-L reagent kit (Diagnostic Chemicals Limited, Canada). The percentage of neuron death was calculated as (LDH activity in cell medium/total LDH content) × 100, where total LDH content was determined after cell lysis by 0.2% Triton X-100 for 15 min at 37°C.
Morphological estimation of neuron death (apoptosis) included the study of nuclear fragmentation using a fluorescent DNA-tropic dye, Hoechst 33342 (absorption wavelength 360 nm, emission wavelength 460 nm), that is able to penetrate into living cells and bind to A-G pairs of damaged fragmented DNA [29]. Stained cells were investigated and visually counted using a fluorescence microscope Axiovert 200 (Zeiss, Germany); the amount of apoptotic cells was expressed as percentage.
Registration of intracellular calcium concentration [Ca2+]i. [Ca2+]i was measured spectrophotometrically using fluorescent probes Fura-2/AM or Fura-2FF/AM with high or low affinity to Ca2+, respectively. The fluorescence spectrum for these lipophilic probes is shifted to lower wavelength upon binding of the probe molecule to Ca2+. Prior to the experiments the cells were loaded with the probe (5 µM) for 40 min in the culture medium, after which the cell culture was washed off with HBSS. A cell-coated glass was placed in a perfusion chamber (0.2 ml) assembled on the table of the Axiovert 200 microscope. Light of two different wavelengths (340 and 380 nm, alternating in 100-200 msec pulses) was used for excitation of fluorescence; emission was assayed at 505 ± 10 nm. A CCD video camera (Roper Scientific, USA) and computer software Metafluor 6.1 (Universal Imaging Corp., USA) allowed obtaining and processing a digital record of the experiment. In the experiments where Fura-2FF/AM was used, the change in [Ca2+]i was estimated by the fluorescence ratio at 340 and 380 nm (F340/F380). In other experiments the absolute values of [Ca2+]i were determined using calibration solutions according to [30]. In all experiments, the background fluorescence was subtracted from registered fluorescence emitted by neurons. All experiments were performed at room temperature (26°C).
Materials. Bovine thrombin, bovine FXa, fluorescent dye Hoechst 33342, NaCl, KCl, CaCl2, MgCl2, KH2PO4, HEPES, glucose, EGTA, 2-aminoethoxydiphenyl borate (2-APB), dantrolene, ionomycin, tetrodoxin, glutamate, NMDA, phenylmethylsulfonyl fluoride (PMSF), glutamax, AraC, Triton X-100, and poly-D-lysine were from Sigma (USA); Fura-2/AM and Fura-2FF/AM from Molecular Probes (USA); synthetic PAR1 agonist peptide (PAR1-AP, TFLLRN) and PAR2 agonist peptide (PAR2-AP, SLIGRL) were from Biosyntan (Germany); PAR1 antagonist (Mpr(Cha)) (Mercaptopropionyl-F(Cha-Cha)RKPNDK-NH2) was kindly provided by A. Kawabata; neurobasal medium A containing 2% Supplement B-27 (Gibco, USA) and L-glutamine (Gibco) was from Gibco; LDH-L reagent kit was from Diagnostic Chemicals Limited.
Statistical treatment of data was performed using Student's t-test for paired samples.
RESULTS
Effect of thrombin and FXa on the death of hippocampal neurons caused by glutamate toxicity. In the first set of experiments the death (by necrosis) of cultured rat hippocampal neurons was studied 24 h after exposure to 100 µM glutamate (for 15 min) or combined effect of glutamate, thrombin, and synthetic peptide - a PAR1 agonist (PAR1-AP) or PAR1 antagonist (Mpr(Cha)) using biochemical techniques (Fig. 1, a and b). Glutamate induced cell death in 34% of hippocampal neurons (Fig. 1a). Incubation of cell culture with thrombin at concentrations from 0.1 to 10 nM resulted in almost two-fold decrease in glutamate-induced neuron death (Fig. 1a). Thrombin at high concentration (100 nM) did not have a protective effect. Moreover, 100 nM thrombin caused cell death in 32% of neurons, which is comparable with glutamate-induced death (data not shown).
Experiments with specific PAR1 agonist peptide (TFLLRN) that selectively activates only PAR1 and with PAR1 antagonist peptide (Mpr(Cha)) indicate the involvement of PAR1 receptors in neuron activation by thrombin. According to our data, PAR1-AP (100 µM) protected neurons from the death from glutamate toxicity similarly to thrombin (Fig. 1b), whereas PAR1 antagonist peptide, Mpr(Cha) (100 µM), prevented the neuroprotective effect of thrombin (Fig. 1b).Fig. 1. Effect of thrombin, synthetic PAR1 agonist peptide (PAR1-AP), and PAR1 antagonist (Mpr(Cha)) on the death of hippocampal neurons induced by incubation of cell culture with 100 µM glutamate for 15 min. Cell death was estimated by the level of lactate dehydrogenase in cell cultures 24 h after application of glutamate. a) Effect of thrombin at concentrations from 0.1 to 100 nM on neuron death; b) effect of PAR1-AP (100 µM) and PAR1 antagonist Mpr(Cha) (100 µM) in combination with thrombin (10 nM) on death of neurons. * p < 0.05 compared to glutamate, here and further n = 5-7 independent experiments.
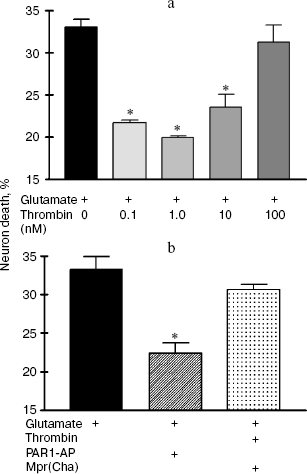
In the second set of experiments, the death of rat hippocampal neurons was studied by morphological techniques using the apoptosis marker Hoechst 33342. Apoptotic neurons had condensed chromatin and fragmented nucleus (Fig. 2, a and b; see color insert). As seen from Fig. 2c, glutamate caused apoptosis after 24 h in 34% of neurons, thrombin (10 nM) decreased the number of apoptotic neurons to 17%, PAR1-AP (100 µM) imitated the neuroprotective effect of thrombin, and PAR1 antagonist peptide (100 µM) prevented the protective effect of thrombin. Therefore, the results of biochemical and morphological studies have confirmed our preliminary data on the involvement of PAR1 in the neuroprotective effect of thrombin [31].
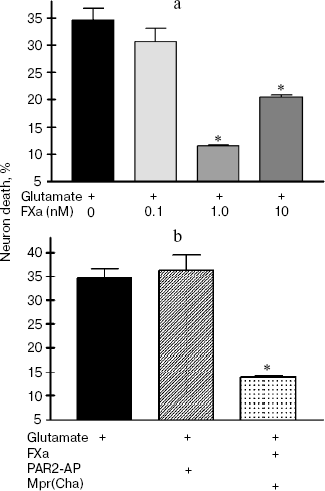
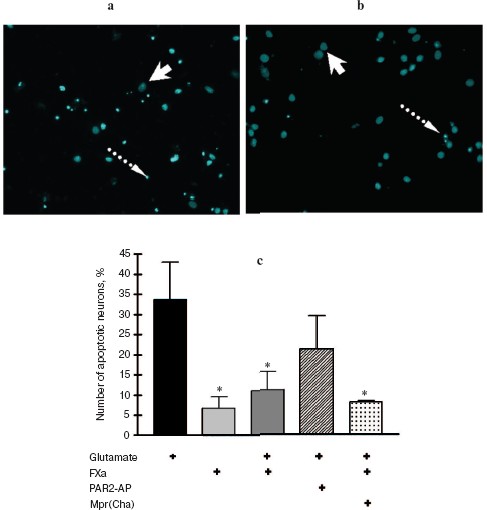
In the next two sets of experiments the influence of a protease, FXa, on glutamate cytotoxicity was studied using the biochemical (Fig. 3) and morphological (Fig. 4, see color insert) approaches. It was found that FXa at the concentration of 1 and 10 nM decreased neuron death caused by a 15 min exposure to 100-µM glutamate down to 12 and 20%, respectively (Fig. 3a). The block of PAR1 receptors by Mpr(Cha) antagonist did not prevent the protective effect of FXa (Figs. 3b and 4c). Since the effect of FXa on cells is mediated predominantly by PAR2 [3], we used a synthetic PAR2 agonist peptide (PAR2-AP) (100 µM) for simulation of the FXa effect on neuron survival after glutamate toxicity. However, as seen from Figs. 3b and 4c, cell death after the incubation of neuron cultures with glutamate and PAR2-AP did not reliably differ from that caused by glutamate alone. The data obtained by different techniques are in good agreement with each other and provide evidence that the PAR1 and PAR2 receptors are not involved in the neuroprotective effect of FXa. Apparently, the protective effect of FXa is realized through a yet unknown subtype of protease-activated receptors, in so far as the presence of proteolytic activity is absolutely necessary for displaying the effect of FXa. We have demonstrated that 10 nM FX, inactivated by PMSF, as well as PMSF-inactivated thrombin (10 nM), did not protect hippocampal neurons from death under conditions of glutamate cytotoxicity (30.5% neuron death vs. 34% resulting from the effect of glutamate alone). These results confirm the data obtained earlier regarding the protective effect of FXa during glutamate cytotoxicity [32].Fig. 2. Effect of thrombin on apoptosis of hippocampal neurons 24 h after the incubation of cell culture with 100 µM glutamate for 15 min. Results of cell count for living and apoptotic neurons stained with nuclear fluorescent dye Hoechst 33243. Solid arrow shows living cells and dashed arrow shows apoptotic cells. a) Cell fluorescence after the effect of glutamate; b) the same after the effect of glutamate in combination with 10 nM thrombin; c) effect of 10 nM thrombin, 100 µM PAR1 agonist (PAR1-AP), and 100 µM PAR1 antagonist (Mpr(Cha)) in combination with thrombin on glutamate-induced apoptosis. * p < 0.05 compared to glutamate.
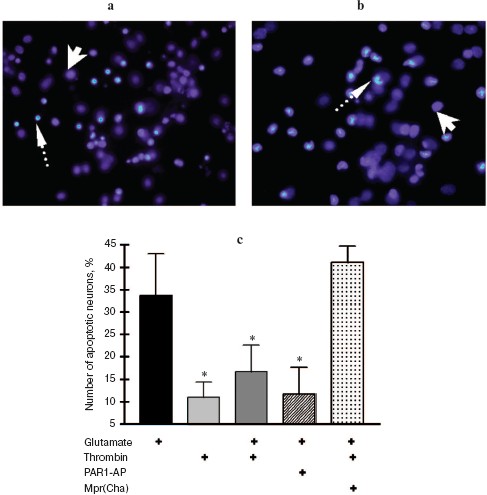
Fig. 3. Effect of factor Xa (FXa), PAR1 antagonist (Mpr(Cha)), and synthetic PAR2 agonist peptide (PAR2-AP) on the death of neurons caused by incubation of cell culture with 100 µM glutamate for 15 min. Cell death was estimated by the level of lactate dehydrogenase in cultures 24 h after application of glutamate. a) Effect of FXa at concentrations from 0.1 to 10 nM on neuron death; b) effect of PAR2-AP (100 µM) and PAR1 antagonist (Mpr(Cha)) (100 µM) in combination with 10 nM FXa on glutamate-induced death of neurons. * p < 0.05 compared to glutamate.
Effect of thrombin and FXa on [Ca2+]i in hippocampal neurons. As seen from Fig. 5, activation of hippocampal neurons by 10 nM thrombin elicited a rapid, transient increase in [Ca2+]i, the concentration of which reached a maximum (150-200 nM) for 20-30 sec. Then, despite the presence of thrombin in the incubation medium, [Ca2+]i reverted to the initial level. Not all hippocampal neurons responded to thrombin by changing [Ca2+]i; the number of responding neurons was on average 64% of the total number of studied neurons (n = 36).Fig. 4. Effect of FXa on apoptosis of hippocampal neurons 24 h after a 15 min incubation of cell culture with 100 µM glutamate. Results of cell count for living and apoptotic neurons stained with nuclear fluorescent dye Hoechst 33243. Solid arrow shows living cells and dashed arrow shows apoptotic cells. a) Cell fluorescence after the effect of glutamate; b) the same after the effect of glutamate in combination with 10 nM FXa; c) effect of 10 nM FXa, 100 µM PAR2 agonist (PAR2-AP), and 100 µM PAR1 antagonist (Mpr(Cha)) in combination with FXa on glutamate-induced apoptosis. * p < 0.05 compared to glutamate.
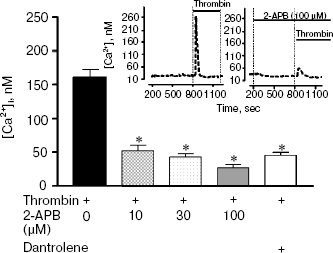
Preliminary incubation of cells with 2-APB (10-100 µM), an IP3 receptor blocker, decreased the calcium response in neurons by more than three-fold (Fig. 5). In such case, thrombin response in the presence of 2-APB was observed in 24% of cells. Incubation of cells with 30 µM dantrolene, an inhibitor of ryanodine receptors, did not change the number of cells responding to thrombin, but decreased the amplitude of calcium response by 3.1-fold. Contrary to thrombin, FXa did not induce the change in [Ca2+]i at all concentrations studied (0.1-80 nM) (data not shown).Fig. 5. Block of IP3 receptors in neuron endoplasmic reticulum by 2-aminoethoxydiphenyl borate (2-APB) (10, 30, and 100 µM) or ryanodine receptors by dantrolene (30 µM) decreases the calcium signal induced by 10 nM thrombin. * p < 0.05 compared to glutamate. Insert at the top shows the original record of [Ca2+]i change in hippocampal neuron in response to thrombin (left) and thrombin in combination with 2-APB (right). [Ca2+]i was measured using the fluorescent probe Fura-2.
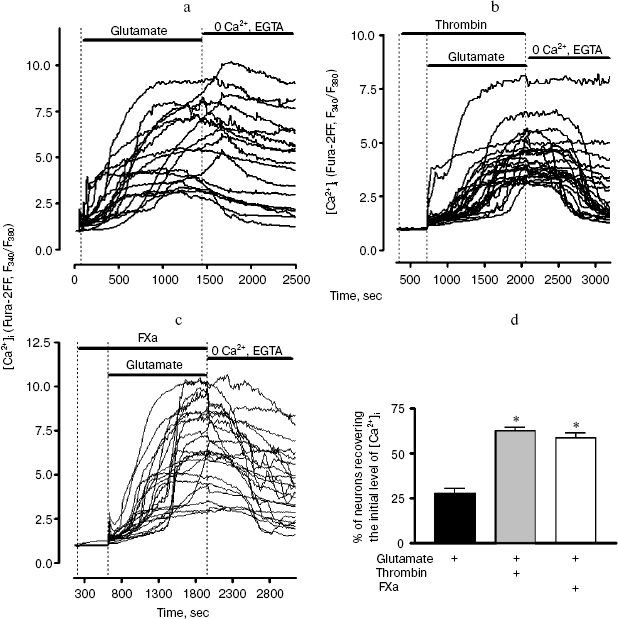
Next we studied the effect of thrombin and FXa on the change in [Ca2+]i (by the ratio of F340/F380) in response to prolonged exposure (20 min) to 100 µM glutamate (Fig. 6). The ability of neurons to regenerate the basal level of [Ca2+]i after the removal of glutamate from the solution has been analyzed (Fig. 6d). In these experiments low affinity probe Fura2-FF, which is sensitive only to substantial increase in [Ca2+]i (more than 1 µM) was used for the registration of [Ca2+]i signal. Glutamate caused a steady two-phase increase in [Ca2+]i in neurons (Fig. 6a). After removal of glutamate, [Ca2+]i level in the vast majority of neurons did not revert to the original level (Fig. 6a). Incubation of cells with 10 nM thrombin did not have a significant effect on the increase in [Ca2+]i caused by glutamate; however, it increased by more than two times the number of neurons in which the basal level of [Ca2+]i was recovered after removal of glutamate (Fig. 6, b and d). Similarly to thrombin, FXa facilitated rapid recovery of intracellular calcium homeostasis after prolonged exposure to glutamate (Fig. 6, c and d).
Fig. 6. Effect of thrombin and FXa on the change in [Ca2+]i (by ratio of F340/F380) in hippocampal neurons upon prolonged exposure to 100 µM glutamate. Low affinity probe Fura-2FF was used. a) The original record of change in [Ca2+]i in individual neurons before, during, and after the effect of glutamate. The cells were washed free of glutamate using calcium-deprived medium containing 100 µM EGTA; b) incubation of cell culture with 10 nM thrombin; c) incubation of cell culture with 10 nM FXa; d) effect of thrombin and FXa on the ability of neurons to recover the basal level of [Ca2+]i after the removal of glutamate. The number of cells (%) in which the F340/F380 ratio 10 min after the washing free of glutamate was started did not exceed the basal level by more than 25% is shown. * p < 0.01 compared to glutamate.
DISCUSSION
It is known that during focal cerebral ischemia the amount of the neurotransmitter glutamate, the toxic effect of which on neurons causes rapid, non-controlled increase in [Ca2+]i, is significantly increased in the intercellular space [33-35]. The disruption of [Ca2+]i homeostasis triggers a cascade of intracellular reactions resulting in rapid or delayed cell death by apoptosis or necrosis [26, 27].
Primary neuron culture is a convenient experimental model for study of neurodestructive processes caused by glutamate, since the cytological and biochemical characteristics of cultured cells are similar to those observed in neurons in situ [36]. The glutamate concentration used in this work (100 µM) had a neurotoxic effect on hippocampal neurons, as indicated by the experiments for estimation of neuron cell death resulting from glutamate exposure (Figs. 1 and 2) as well by change in [Ca2+]i (Fig. 6a). Hence, the selected model of glutamate cytotoxicity is comparable to endogenous pathological conditions of the injured brain.
In recent times, interest in the effect of serine proteases of the blood coagulation system beyond homeostasis has dramatically increased [3, 4, 6, 10]. However, the data on the effect of hemostasis proteases on neurons are contradictory and not abundant. We have attempted to characterize the role of thrombin, a key serine protease of hemostasis, and its precursor in the coagulation cascade, FXa, using the model of glutamate cytotoxicity in cultured rat hippocampal neurons.
The data indicate that at low concentrations (up to 10 nM) thrombin and FXa display a neuroprotective effect. At higher concentrations (100 nM), these proteases cause cell death. The minimal neuroprotective concentration of thrombin is 0.1 nM, and the corresponding FXa concentration is one order of magnitude higher, 1 nM (Fig. 3a). Different enzyme specificity to their substrates, PAR receptors, can be explained by a certain difference in the tertiary structure of these related enzymes. In the thrombin molecule there is a specific anion-binding exosite ABE1, which recognizes a complementary region in PAR1 exodomain, which in turn is similar to the negatively charged C-terminus of hirudin, a highly specific thrombin inhibitor [37]. Structural features of the enzymes are responsible for the difference between thrombin and FXa in the choice of their substrates and receptors. Our data on the protective effect of low thrombin concentrations confirm the results of a previous investigation [38], where the effect of thrombin on neurons was studied using hippocampal slices for modeling ischemia; however, they are not in agreement with other data [39] on the ability of thrombin to potentiate NMDA receptors in hippocampal neurons.
The effect of thrombin on cells is mediated by PAR1 as well as PAR4 [3, 4, 13, 40]. The use of agonist and antagonist showed that thrombin imparts a neuroprotective effect on hippocampal neurons through PAR1, since the PAR1 antagonist prevented the protective effect of thrombin, whereas the PAR1 agonist imitated its effect (Figs. 1b and 2c). Thus, thrombin exerts its neuroprotective effect in a narrow concentration range (0.1-10 nM) in conjunction with PAR1 receptors. It is probable that the destructive effect of thrombin simultaneously involves different types of receptors and in large amount.
There is evidence that FXa activates vascular endothelial cells through PAR1 and PAR2 receptors [41]. FXa induced calcium signal in fibroblasts through PAR1 receptors and stimulated cell proliferation and procollagen synthesis [42]. In our experiments, the preliminary incubation of cells with PAR2 agonist peptide did not protect the cells from glutamate-induced death, and with PAR1 antagonist it did not prevent the neuroprotective effect of FXa (Figs. 3b and 4c).
Therefore, according to our data, thrombin displays a protective effect on hippocampal neurons during glutamate toxicity by activation of PAR1. FXa exerts a protective effect through an unknown subtype of PAR, because the effect of FXa is not imitated by the PAR2 agonist and not blocked by PAR1 antagonist (Figs. 3 and 4).
Since glutamate at toxic concentrations causes irreversible increase in [Ca2+]i, it can be assumed that the reduction of neurotoxic glutamate effect on cells by thrombin is mediated by modulation of [Ca2+]i. There are sporadic data in literature concerning the change in [Ca2+]i in rat hippocampal neurons caused by thrombin [43]. In the present work we have confirmed the data obtained by us earlier [31] that indicated that PAR1 agonist peptide imitates the thrombin-induced calcium signal in hippocampal cells, whereas the antagonist blocks it. It was demonstrated that PAR1 is involved in the calcium signal induced by thrombin in human brain astrocytes [44].
Increase in [Ca2+]i in neurons may be associated both with mobilization of Ca2+ from intracellular stores and Ca2+ influx into cells. Earlier we have shown [31] that in calcium-free medium (100 µM EGTA), thrombin causes an increase in [Ca2+]i, but the amplitude of calcium signal is more than two times lower than in calcium-containing medium. After depleting the reticulum of neurons in calcium-free medium using cyclopiazonic acid (an inhibitor of reticulum Ca2+-ATPase), the addition of thrombin did not result in increase in [Ca2+]i. These data indicate the role of intracellular reticulum in thrombin-induced calcium signal. Release of calcium from the reticulum can be realized by activation of two types of receptors, IP3 and ryanodine. The use of corresponding blockers of the above receptors, 2-ABP and dantrolene, showed the predominant role of the IP3 receptors in the calcium response to thrombin (Fig. 5). These results are in agreement with the data illustrating that the mechanism of PAR1-dependent calcium response in oligodendrocytes is also mediated by the IP3 receptors of endoplasmic reticulum [45]. However, some authors mention that the reticulum IP3 receptors are involved in the induction of death of granule cells in cerebellum [46].
Since the apoptotic effect of glutamate is based on the overloading of cells by calcium and disruption in the regulation of calcium homeostasis, we have studied the effect of thrombin on glutamate-induced increase in [Ca2+]i in hippocampal neurons. It has been demonstrated that the incubation of cell culture with thrombin at the concentration of 10 nM or with FXa at the same concentration improves the recovery of calcium homeostasis in neurons after exposure to glutamate (Fig. 6). The mechanism of the effect is unknown; however, it can be assumed that through the activation of the intracellular factors serine proteases facilitate the calcium transport system (first of all, Ca2+-ATPase of the plasma membrane), resulting in the enhancement of calcium efflux. It cannot be excluded that proteases might diminish the [Ca2+]i signal induced by activation of glutamate receptors. Thus, we have previously found that preliminary incubation of cells with low concentrations of thrombin resulted in a decrease in intracellular calcium signal in hippocampal neurons upon application of NMDA, an agonist of NMDA subtype of glutamate receptor [31]. According to literature data [47], neuroprotection in hippocampal neurons can be achieved through decrease in ionic current mediated by NMDA receptors. At the same time, we have not found any change in [Ca2+]i in hippocampal neurons induced by FXa. However, both thrombin and FXa decreased the number of hippocampal neurons in which the prolonged exposure to glutamate induced irreversible increase in [Ca2+]i (Fig. 6). This matter requires further investigation.
As pointed out above, thrombin is able to activate PAR1 and PAR4 [17, 40]. However, the PAR4 molecule lacks the site complementary to anion-binding exosite 1 (ABE1) in thrombin, which is responsible for binding of highly specific substrates and receptors, and requires high thrombin concentrations for its activation [3]. This explains heterodirected effects of thrombin. Thus, effect of thrombin on microglia is realized mainly through PAR4, which is involved in brain inflammation processes associated with neurotrauma and neurodegeneration [16, 40]. Thrombin can release NO in rat microglial cell culture, and this being the case, PAR1 inhibitor does not block the above effect [48].
Both proapoptotic and antiapoptotic effects of thrombin are described in the literature. Upon intraventricular introduction, thrombin at high concentrations (25 and 100 nM, 0.25 µl/h, 28 days) caused apoptosis and decreased cognitive function in rats [49]. At concentrations higher than 25 nM, thrombin increased the sensitivity of cultured hippocampal neurons to glucose deprivation [44] and induced apoptosis in dopaminergic neurons [50]. In addition, thrombin protected astrocytes and hippocampal neurons from death caused by hypoglycemia and oxidative stress [38, 51-53].
In summary, though thrombin itself can induce death in a certain small population of hippocampal neurons, upon combined effect (at low concentration) with glutamate it is able to protect cells from glutamate-induced apoptosis and, moreover, a long time after the exposure to glutamate. The mechanism of neuroprotection is unknown. Activation of G-protein-coupled PAR1 receptors causes hydrolysis of phosphoinositides with formation of diacylglycerol, a well-known protein kinase C activator [3, 4, 6]. Perhaps the transduction of intracellular signal upon PAR1 activation occurs by the same pathway as for the activation of metabotropic glutamate receptors, which are also G-protein coupled. It has been shown that preliminary incubation of these receptors decreases the subsequent toxic effect of glutamate [54]. The increase in intracellular Ca2+ concentration induced by activation of PAR can influence a number of intracellular factors, both in nucleus and cytoplasm (transcription factors NF-kappaB, FosB/JunD, AP-1, and others) and trigger/suppress the expression of different genes including those proteins (Bax/Bad from Bcl family, AIF, MAP kinases, caspases) whose activity determines the progression of apoptosis during the toxic effect of glutamate [26, 55]. Investigation of mechanisms of interaction between glutamate and PAR receptors is a promising direction in the search for new approaches to treatment of cerebrovascular diseases.
This work was supported by the Russian Foundation for Basic Research grants (Nos. 05-04-49481 and 04-04-48513).
REFERENCES
1.Coughlin, S. R. (1999) Proc. Natl. Acad. Sci.
USA, 96, 11023-11027.
2.Coughlin, S. R. (2000) Nature, 407,
258-264.
3.Ossovskaya, V. S., and Bunnett, N. W. (2004)
Physiol. Rev., 84, 579-621.
4.Strukova, S. M. (2001) Biochemistry
(Moscow), 66, 8-18.
5.Derian, C. K., Demiano, B. P., D'Andrea, M. R., and
Andrade-Gordon, P. (2002) Biochemistry (Moscow), 67,
56-64.
6.Macfarlane, S. R., Seatter, M. J., Kanke, T.,
Hunter, G. D., and Plevin, R. (2001) Pharmacol. Rev., 53,
245-82.
7.Strukova, S. M., Serejskaya, A. A., and Osadchuk,
T. V. (1989) Uspekhi Sovrem. Biol., 107, 41-54.
8.Sheehan, J. P., and Sadler, J. E. (1994) Proc.
Natl. Acad. Sci. USA, 91, 5518-5522.
9.Stubbs, M., and Bode, W. (1995) Trends Biochem.
Sci., 20, 23-28.
10.Balezina, O. P., Gerasimenko, N. Yu., Dugina, T.
N., and Strukova, S. M. (2004) Uspekhi Fiziol. Nauk, 35,
37-49.
11.Suo, Z., Citron, B. A., and Festoff, B. W.
(2004) Curr. Drug Targets Inflamm. Allergy, 3,
105-114.
12.Striggow, F., Riek, M., Breder, J.,
Henrich-Noack, P., Reymann, K. G., and Reiser, G. (2000) Proc. Natl.
Acad. Sci. USA, 97, 2264-2269.
13.Striggow, F., Riek-Burchardt, M., and Reiser, G.
(2001) Eur. J. Neurosci., 14, 595-608.
14.Yoshida, S., and Shiosaka, S. (1999) Int. J.
Mol. Med., 3, 405-409.
15.Riek-Burchardt, M., Striggow, F., Henrich-Noack,
P., Reiser, G., and Reymann, K. G. (2002) Neurosci. Lett.,
329, 181-184.
16.Xi, G., Reiser, G., and Keep, R. F. (2003) J.
Neurochem., 84, 3-9.
17.Wang, H., Ubl, J. J., and Reiser, G. (2002)
Glia, 37, 53-63.
18.Pompili, E., Nori, S. L., Geloso, M. C.,
Guadagni, E., Corvino, V., Michetti, F., and Fumagalli, L. (2004)
Brain Res. Mol. Brain Res., 122, 93-98.
19.Rohatgi, T., Henrich-Noack, P., Sedehizade, F.,
Goertler, M., Wallesch, C. W., Reymann, K. G., and Reiser, G. (2004)
J. Neurosci. Res., 75, 273-279.
20.Smith-Swintosky, V. L., Cheo-Isaacs, C. T.,
D'Andrea, M. R., Santulli, R. J., Darrow, A. L., and Andrade-Gordon, P.
(1997) J. Neurochem., 69, 1890-1896.
21.Kawabata, A., Oono, Y., Yonezawa, D., Hiramatsu,
K., Inoi, N., Sekiguchi, F., Honjo, M., Hirofuchi, M., Kanke, T., and
Ishiwata, H. Br. (2005) J. Pharmacol., 144, 212-219.
22.Noorbakhsh, F., Vergnolle, N., McArthur, J. C.,
Silva, C., Vodjgani, M., Andrade-Gordon, P., Hollenberg, M. D., and
Power, C. J. (2005) Immunol., 174, 7320-7329.
23.Jin, G., Hayashi, T., Kawagoe, J., Takizawa, T.,
Nagata, I., Syoji, M., and Abe, K. J. (2005) Cerebr. Blood Flow
Metab., 25, 302-313.
24.Olson, E. E., Lyuboslavsky, P., Traynelis, S. F.,
and McKeon, R. J. (2004) J. Cerebr. Blood Flow Metab.,
24, 964-971.
25.Hossain, M. A. (2005) Epilepsy Behav.,
7, 204-213.
26.Orrenius, S., Zhivotovsky, B., and Nicotera, P.
(2003) Nat. Rev. Mol. Cell Biol., 4, 552-565.
27.Dingledine, R., Borges, K., Bowie, D., and
Traynelis, S. F. (1999) Pharmacol. Rev., 51, 7-60.
28.Khodorov, B. I., Storozhevykh, T. P., Surin, A.
M., Sorokina, E. G., Yuryavichus, A. I., Borodin, A. V., Vinskaya, N.
P., Khaspekov, L. G., and Pinelis, V. G. (2001) Biol. Membr.
(Moscow), 18, 421-432.
29.Liu, X. Z., Xu, X. M., Hu, R., Du, C., Zhang, S.
X., McDonald, J. W., Dong, H. X., Wu, Y. J., Fan, G. S., Jacquin, M.
F., Hsu, C. Y., and Choi, D. W. (1997) J. Neurosci., 17,
5395-5406.
30.Kiedrowski, L., Brooker, G., and Costa, E. (1994)
Neuron, 12, 295-300.
31.Kiseleva, E. V., Storozhevykh, T. P., Pinelis, V.
G., Gluza, E., and Strukova, S. M. (2004) Byul. Eksp. Biol.
Med., 137, 519-523.
32.Strukova, S., Gorbacheva, L., Storozhevykh, T.,
Pinelis, V., and Smirnov, M. (2006) J. Thromb. Haemost.,
6, 1409-1410.
33.Choi, D. W. (1992) J. Neurobiol.,
23, 1261-1276.
34.Gusev, E. I., Skvortsova, V. I., and Raevsky, K.
S. (1997) Eur. J. Neurol., 4, 78.
35.Danysz, W., and Parsons, C. (1998) Pharmacol.
Rev., 50, 597-664.
36.Khaspekov, L. G. (1995) Mechanisms and Factors
of Neurodestructive Effect of Excitatory Amino Acids on Brain Neurons
in vitro: Author's abstract of Doctoral dissertation [in Russian],
Moscow.
37.Gaussem, P., Picard, V., Chadeuf, G., Arnaud, E.,
and Aiach, M. (1995) FEBS Lett., 365, 219-222.
38.Henrich-Noack, P., Striggow, F., Reiser, G., and
Reymann, K. G. (2005) J. Neurosci. Res., 83, 128-133.
39.Gingrich, M. B., Jung, C. E., Lyuboslavsky, P.,
and Traynelis, S. F. (2000) J. Neurosci., 20,
4582-4595.
40.Suo, Z., Wu, M., Citron, B. A., Gao, C., and
Festoff, B. W. (2003) J. Biol. Chem., 278,
31177-31183.
41.McLean, K., Schirm, S., Johns, A., Morser, J.,
and Light, D. R. (2001) Thromb. Res., 103, 281-297.
42.Blanc-Brude, O. P., Archer, F., Leoni, P.,
Derian, C., Bolsover, S., Laurent, G. J., and Chambers, R. C. (2005)
Exp. Cell Res., 304, 16-27.
43.Smith-Swintosky, V. L., Zimmer, S., Fento, J. W.,
II, and Mattson, M. P. (1995) J. Neurosci., 15,
5840-5850.
44.Junge, C. E., Lee, C. J., Hubbard, K. B., Zhang,
Z., Olson, J. J., Hepler, L. R., Brat, D. J., and Traynelis, S. F.
(2004) Exp. Neurol., 188, 94-103.
45.Wang, Y., Richter-Landsberg, C., and Reiser, G.
(2004) Neuroscience, 126, 69-82.
46.Limke, T. L., Bearss, J. J., and Atchison, W. D.
(2004) Toxicol. Sci., 80, 60-68.
47.Blaabjerg, M., Baskys, A., Zimmer, J., and
Vawter, M. P. (2003) Mol. Brain Res., 117, 196-205.
48.Ryu, J., Pyo, H., Jou, I., and Joe, E. J. (2000)
Biol. Chem., 275, 29955-29959.
49.Mhatre, M., Nguyen, A., Kashani, S., Pham, T.,
Adesina, A., and Grammas, P. (2004) Neurobiol. Aging, 25,
783-793.
50.Choi, S. H., Lee, da Y., Pyu, J. K., Kim, J.,
Joe, E. H., and Jin, B. K. (2003) Neurobiol. Dis., 14,
181-193.
51.Vaughan, P. J., Pike, C. J., Cotman, C. W., and
Cunningham, D. D. (1995) J. Neurosci., 15, 5389-5401.
52.Chalmers, C. J., Balmanno, K., Hadfield, K., Ley,
R., and Cook, S. J. (2003) Biochem. J., 375, 99-109.
53.Strukova, S. M., Kiseleva, E. V., Dugina, T. N.,
Gluza, E., Storozhevykh, T. P., and Pinelis, V. G. (2005) Fiziol.
Zh. im. I. M. Sechenova, 91, 53-60.
54.Sanchez-Perez, A., Llansola, M., Cauli, O.,
and Felipo, V. (2005) Cerebellum, 4, 162-170.
55.Flynn, A. N., and Buret, A. G. (2001)
Apoptosis, 9, 729-737.