Random Mutagenesis of Luciola mingrelica Firefly Luciferase. Mutant Enzymes with Bioluminescence Spectra Showing Low pH Sensitivity
M. I. Koksharov and N. N. Ugarova*
Department of Chemical Enzymology, Faculty of Chemistry, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-2660; E-mail: nugarova@gmail.com* To whom correspondence should be addressed.
Received December 29, 2007; Revision received April 22, 2008
Most firefly luciferases demonstrate a strong pH-dependence of bioluminescence spectra. Gene region encoding first 225 residues of Luciola mingrelica luciferase was subjected to random mutagenesis, and four mutants with altered pH-sensitivity of bioluminescence spectra were isolated. F16L substitution showed distinctly lower pH-dependence of bioluminescence spectra, and Y35N,H and F16L/A40S substitutions resulted in the enzymes with bioluminescence spectra virtually independent from pH in the range of 6.0-7.8. The structural explanation is proposed for the effect of mutations on pH-sensitivity of bioluminescence spectra.
KEY WORDS: firefly luciferase, random mutagenesis, mutation of amino acid residues, bioluminescence spectra, pH dependence of bioluminescence spectraDOI: 10.1134/S0006297908080038
Abbreviations: DLSA) 5´-O-[N-(dehydroluciferyl)-sulfamoyl]adenosine; DTT) dithiothreitol; E) luciferase; LO) oxyluciferin; PCR) polymerase chain reaction; lambdamax) maximum of bioluminescence spectra.
Firefly luciferase (EC 1.13.12.7) catalyses the oxidation of luciferin
by oxygen in air in the presence of ATP and Mg2+ [1]. The enzyme transfers the energy of the chemical
reaction into light with high efficacy [2, 3]. Due to high catalytic activity, ATP specificity,
and simple registration of the bioluminescent signal, firefly
luciferase is widely used for ATP determination and as a reporter gene
in studies of different biochemical processes [4,
5]. The maximum of the bioluminescence spectrum
(lambdamax) of luciferases isolated from different
beetles varies from 536 to 623 nm [6]. Recently a
firefly was reported with lambdamax of 504 nm
[7], but the amino acid sequence of the
corresponding luciferase is not known yet. The chemical mechanism of
the reaction and the product structure are identical for all beetle
luciferases, and the properties of luciferase determine differences in
bioluminescence spectra [8]. Beetle luciferases can
be divided in two groups according to the sensitivity of their
bioluminescence spectra to pH [6]: i)
pH-insensitive luciferases from click-beetles and some other species
with lambdamax and spectral shape virtually unchanged
upon decreasing pH, ii) pH-sensitive firefly luciferases, whose
lambdamax shifts from 540-570 nm (green or
yellow-green light) at pH 8.0 to ~620 nm (red light) at pH =
6. This significant shift is usually explained by the assumption that
the reaction product (electron-excited oxyluciferin) can exist in the
active center in two different molecular forms--“green” and
“red” emitters. Their ratio is different under different
conditions, thus determining the lambdamax and shape
of the bioluminescence spectrum. The specific nature of these forms as
well as the mechanism of different pH sensitivities of luciferases is
still the subject of discussions [6, 9-12]. A number of mutations
leading to decrease in pH sensitivity of bioluminescence spectra of
firefly luciferases are described in the literature. In some cases, the
cumulative effect of substitution of several amino acid residues led to
emission spectra that did not vary in the range of pH from 6 to 8 [13, 14].
Mutations affecting the pH dependence of bioluminescence spectra are virtually unknown for the first 225 amino acid residues of luciferase. It is considered that this part of the enzyme molecule does not play a significant role in the definition of bioluminescence color. In the present work, random mutagenesis of this region of Luciola mingrelica firefly luciferase was performed, and a number of mutant enzymes with bioluminescence spectra different from those for the native luciferase and apparently insensitive to pH were obtained. Properties of these mutant enzymes were investigated.
MATERIALS AND METHODS
The following reagents were used in the present work: restrictases NheI and BamHI (Fermentas, Lithuania); AsuNHI, Pfu DNA-polymerase (Sibenzyme, Russia); dNTPs, T4 DNA-ligase (USB, USA); Taq DNA-polymerase (Sileks, Russia); ATP disodium salt, dithiothreitol (DTT) (ICN, USA). Firefly luciferin was synthesized at the Department of Chemical Enzymology at Lomonosov Moscow State University [15]. Other chemicals used were analytical or chemical grade. Competent cells were obtained and plasmid transformation was performed according to a described method [16].
DNA sequencing was performed using an ABI PRISM → BigDye . Terminator v.3.1 kit with the subsequent analysis of the products on an ABI PRISM 3100-Avant automatic DNA sequencer.
Plasmids used. Three additional restriction sites in the region between NheI-BamHI were introduced in the initial plasmid, pLR [17], encoding firefly luciferase from L. mingrelica, by overlap extension PCR method [18]. These were (the corresponding amino acid and its codon replacement are specified in the parentheses): BstBI (S44, AGT/TCG); SalI (V50, GTT/GTC); XhoI (S130, CCC/TCG). The thus encoded amino acid sequence of luciferase was not changed. The resulting pLR3 plasmid was used for the generation of mutants. Plasmid pETL4 used for the luciferase expression in E. coli was obtained as follows. The region between NcoI-NheI sites in pET23b vector (Novagen, USA) was replaced with the corresponding region from pET28a vector (Novagen), and the luciferase gene from pLR3 was inserted between the NheI site and the transcription terminator. Thus, pETL4 encodes luciferase containing an additional 24 amino acid residues at the N-terminus including 6xHis-tag: MGSSHHHHHHSSGLVPRGSHMASK.
Random mutagenesis and preparation of the mutant library. Random mutagenesis of the L. mingrelica luciferase gene in the region between NheI and BamHI sites (680 bp) was performed by error-prone PCR [19]. Forward and reverse primers used were respectively: 5´-ATTATAGGAGGCTAGCAAAATGG-3´ and 5´-CGTAAATTGGATCCTTAGCGTG-3´. The reaction mix (50 µl) contained 10 mM Tris-HCl (pH 8.3 at 25°C), 50 mM KCl, 7 mM MgCl2, 0.2 mM MnCl2, 0.2 mM dATP, 0.2 mM dGTP, 1 mM dCTP, 1 mM dTTP, 20 pmol of each primer, ~2 fmol of pLR3, and 2.5 units of Taq DNA-polymerase. Polymerase chain reaction was performed on a Tercik amplifier (DNA-Technology, Russia) under the following conditions: 95°C, 1 min; 25 cycles for 1 min at 94°C, 1 min at 53°C, 1 min at 72°C; then 10 min at 72°C. PCR products were resolved on 1% agarose gel with subsequent isolation from the gel using a Qiagen kit. The fragment obtained was treated with restrictases NheI and BamHI, purified from low molecular weight restriction products by electrophoresis, isolated from the gel, and ligated into pLR3 plasmid cut at the same sites, thus preparing a mix of plasmids containing mutant luciferase genes. Escherichia coli strain XL1blue was transformed with the mix, and cells were plated on Petri dishes with LB medium containing 100 µg/ml ampicillin.
Mutant library screening according to brightness and color of bioluminescence. Dishes obtained during the primary screening as described above were incubated overnight at 37°C and then for 6-8 h at room temperature. In vivo luminescence of colonies was registered according to the protocol from [20]: dishes were filled with solution of 1 mM luciferin in 0.1 M Na-citrate buffer (pH 5.0), shaken in the dark, and photographed with a Canon PowerShot A530 camera. Five hundred to 3500 colonies were screened on a Petri dish (diameter 90 mm) according to this protocol. The most interesting colonies from the point of view of color and brightness were transferred during the secondary screening onto two dishes with LB medium containing 100 µg/ml of ampicillin. Several colonies expressing native luciferase were transferred onto the same dishes for comparison. Cells were grown overnight at 37°C, 6-8 h at room temperature, and the colonies were screened a second time according to the color and brightness of their bioluminescence. The most interesting mutants were grown in a small volume of cell culture and cells were harvested by centrifugation. The pellet was resuspended in 0.1 M Na-phosphate buffer, pH 7.8, containing 2 mM EDTA, 1 mM DTT, 1% neonol, 0.5% Triton X-100, and 1 mg/ml lysozyme, and incubated for 1.5-2 h at 0°C. Cell walls were precipitated by centrifugation, and the bioluminescence spectrum of the lysate was registered at pH 7.8 and 6.0. Based on the analysis of the spectra, mutant luciferase forms were selected for further studies.
Preparation of highly purified luciferase. To prepare the native and mutant luciferases containing 6xHis-tag, corresponding DNA regions from pLR3 plasmid encoding the enzymes were recloned on NheI-BamHI sites in pETL4 plasmid. Plasmids obtained were used to transform E. coli strain BL21(DE3)CodonPlus (Stratagene, USA). Enzyme was prepared according to the auto-induction lactose method [21]. BL21(DE3)CodonPlus cells containing pETL4 plasmid were plated on a dish with LB medium containing 100 µg/ml ampicillin and 1.5% Bacto-agar and incubated overnight at 37°C. Three milliliters of LB with 100 µg/ml ampicillin and 1% glucose were inoculated with several colonies, cells were grown for 5-6 h on a shaker at 37°C, 180 rpm, until the cell suspension became turbid (A600 = 0.5-1.0). Flasks with 200 ml of ZYP-5052 medium [21] containing 100 µg/ml ampicillin were inoculated with 0.8 ml of cell culture and incubated on a shaker for 2 h at 37°C, 180 rpm, until the suspension was slightly turbid (A600 = 0.2-0.8). Then cells were grown for 14-16 h at 23°C until A600 = 5-8. Cells were harvested by centrifugation (5500g, 10 min, 4°C). The pellet was resuspended in 18-20 ml of 20 mM Na-phosphate buffer containing 0.5 M NaCl, pH 7.5 (buffer HB), with 20 mM imidazole and 0.5% Triton X-100 added, sonicated (6 cycles for 30 sec, 1 min intervals), and pelleted (39,000g, 30 min, 4°C). To purify luciferase by metal chelate affinity chromatography, supernatant (~20 ml) was loaded on a 1 ml Ni-IDA column (Amersham, Sweden) and washed with 20-40 ml HB buffer containing 20 mM imidazole. The enzyme was eluted with HB buffer containing 300 mM imidazole. Chromatography was performed at 4°C. Luciferase solution obtained was supplemented with EDTA (0.5 M, pH 8.0) to 2 mM and DTT (1 M in 10 mM Na-acetate buffer, pH 5.2) to 1 mM. This solution did not loose activity for two weeks when stored at 0-4°C. Luciferase was transferred in the following buffer: 50 mM Tris-acetate, 100 mM Na2SO4, 2 mM EDTA, pH 7.3 (25°C) for imidazole removal and for prolonged storage by gel filtration on a K9/20 Sephadex G-25 (Pharmacia, Sweden) column. Fractions were supplemented with DTT to 1 mM and stored at -80°C. Luciferase concentration was determined spectrophotometrically by registering absorbance at 280 nm.
Luciferase activity was determined on an FB12 luminometer (Zylux, USA) using the maximal intensity of the light emitted during the enzymatic reaction at saturating concentrations of substrates. The cuvette contained 0.4 ml of 0.05 M Tris-acetate buffer (2 mM EDTA, 10 mM MgSO4, pH 7.8), 0.3 ml of 4 mM ATP solution in the same buffer, and 10 µl of luciferase solution. Then 0.3 ml of 1 mM luciferin in the same buffer was injected and the intensity of bioluminescence was registered. Activity was expressed in relative light units (RLU) of the luminometer (1 RLU = 2·105 quanta/sec).
Bioluminescence spectra were registered on an LS 50B spectrofluorometer (Perkin-Elmer, USA) in bioluminescence mode at slit width 5 nm. The fluorometric cuvette was filled with 1 ml of 0.05 M Tris-acetate buffer with required pH (5.8-9.5) containing 10 mM MgSO4, 2 mM EDTA, 0.33 mM luciferin, and 1.33 mM ATP, then the concentrated enzyme solution was added (0.5-2% v/v), quickly mixed, and bioluminescence spectra were registered in the range of 480-680 nm. The cuvette in the spectrofluorometer was thermostatted at 25°C if not indicated otherwise. Spectra were corrected for PMT sensitivity using the “Perkin-Elmer FL WinLab” program. Spectra recorded after the intensity drop during the measurement did not exceed 5% were used for the analysis.
Kinetics of thermal inactivation of luciferase was studied incubating the enzyme solution (0.01 mg/ml) in 50 mM Tris-acetate buffer containing 20 mM MgSO4, 2 mM EDTA, pH 7.8, at 42°C. Aliquots were collected at certain time points, incubated for 15 min on ice, and then the enzyme activity was determined.
Computer modeling and analysis of L. mingrelica luciferase structure. Homologous modeling of complexes of L. mingrelica firefly luciferase was based on the structures of luciferase-DLSA (5´-O-[N-(dehydroluciferyl)-sulfamoyl]adenosine) complexes (2D1S) and luciferase-LO-AMP complexes (2D1R) obtained for L. cruciata luciferase (~81% homology with L. mingrelica luciferase) in work [22]. Structures were downloaded from the Protein Data Bank [23]. Server What IF was used for the development of a primary model [24]. A loop region formed by residues 183-189 in L. mingrelica luciferase contains an insert, K188, compared to the homologous fragment of L. cruciata luciferase. Since the What IF server does not allow modeling inserts, the mentioned fragment was modeled by means of the ModLoop server [25]. The structures obtained were used for microenvironment analysis and for the possible effects mutations in the studied amino acid residues of L. mingrelica luciferase. Multiple alignments of luciferase amino acid sequences were performed using the ClustalW algorithm [26].
RESULTS AND DISCUSSION
Random mutagenesis and screening of colonies for mutant luciferases with altered bioluminescence spectrum. The region of the gene between NheI and BamHI restriction sites (680 bp), encoding the first 225 of 548 amino acid residues of firefly luciferase from L. mingrelica, was subjected to mutagenesis. According to the literature [27], under the PCR conditions used in this work it should yield about 2-3 base replacements per mutated region, which corresponds to the replacement of one amino acid residue on average. Such frequency is optimal for random mutagenesis and usually corresponds to 50-60% active mutants in the library [19]. Frequency of the mutagenesis is varied by changing Mn2+ concentration. About 50% active mutants were observed at the concentration used in this work (0.2 mM). Though luciferase generates green luminescence in vitro at the pH optimum of the activity, yellow luminescence was observed in vivo for colonies in E. coli expressing native luciferase. This was apparently caused by lower intracellular pH under the cultivation conditions used, which led to an increase in the red component in the bioluminescence spectrum of the luciferase. During mutant screening a number of colonies with green and reddish bioluminescence were found. Thus, in this case more green colonies were formed by the mutants with an increased stability of the spectrum to change in pH. During primary screening about 3000 colonies were analyzed, and 23 mutants with the brightest luminescence and different bioluminescence color from the initial enzyme were selected. During secondary screening, the color and the brightness of colonies of these mutants were compared under identical growth conditions. Bioluminescence spectra were registered for the most promising mutants at pH 7.8 and 6.0 using cell lysates. Mutants MT2, MT3, and MT4 with bioluminescence spectra least sensitive to pH change and colony brightness higher than that of the initial luciferase were revealed and selected for further studies. Mutant MT8, which had brighter colonies than that of an initial enzyme, was used for repeated mutagenesis resulting in mutant MT6 having low pH sensitivity of the bioluminescence spectrum. Sequencing was used to define replacements in plasmids encoding selected mutants. The table shows the corresponding replacements of amino acid residues and the bioluminescence color of the E. coli colonies expressing these mutant luciferases.
Properties of mutant luciferases
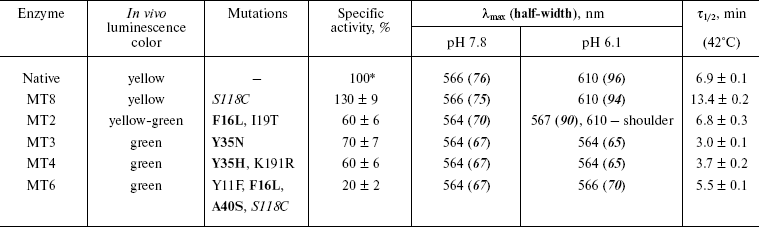
Note: Mutations responsible for change in luciferase bioluminescence
spectra are shown in bold. Half-width, width of spectrum at 50%
height.
*Specific activity of native luciferase was
1.1·1011 RLU/mg protein.
Therefore, four mutants with lower pH sensitivity of the bioluminescence spectrum were obtained by the method of random mutagenesis. Unlike the initial luciferase, the form of their spectra and lmax of bioluminescence did not or only slightly depended on pH in the interval of 6.0-7.8.
Kinetic properties and thermostability of the mutant enzymes. The mutant luciferases have been isolated in highly purified form. The activity of the mutants was from 20 to 130% (mutants MT6 and MT8, respectively) of that for the wild type luciferase (table). Bell-shaped dependence of the activity upon pH with maximum at 7.8-8.0 was conserved for all mutant enzymes as well as for the native enzyme. Thermal inactivation studies at 42°C showed that this process is well described by a single exponential; therefore, the luciferase thermostability was characterized by the time of 50% decrease in activity (tau1/2) (table). Enzyme MT8 containing the replacement S118C had increased thermostability. This residue is located inside the protein globule, its environment is mainly hydrophobic, but it also forms a hydrogen bond with residue N199 and there is a neighboring water molecule. It could be assumed that the replacement of a hydrophilic internal Ser residue with more hydrophobic Cys increases the stability of the internal hydrophobic packing of the protein and thus increases the thermostability of the molecule. The thermostability of enzyme MT2 was the same as for the initial luciferase, and other enzymes were even somewhat less stable. Thermostability decrease for mutants MT3 and MT4 with Y35N and Y35H replacements also correlates with hydrophobicity: the stability decreases when internal hydrophobic Tyr is replaced with polar His or Asn residues. It is likely that decrease in the thermostability of the MT6 mutant in comparison with its initial mutant MT8 is caused by the replacement of internal hydrophobic Ala40 with the larger hydrophilic Ser residue.
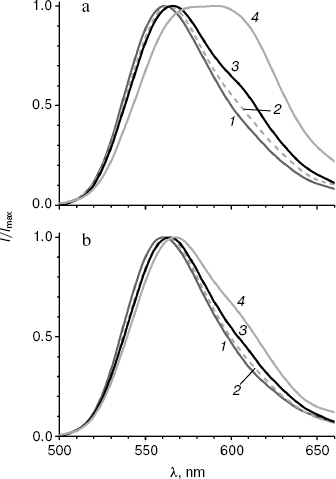
Spectral properties of the mutant enzymes. Bioluminescence spectra were registered for the initial and mutant luciferases in the pH range 5.8-9.5. In the pH region 7.8-9.5, there were almost no change in the shape of spectra and lmax value for all the studied enzymes. Spectral characteristics for the mutants at pH 7.8 and 6.1 are presented in the table. Figure 1 shows bioluminescence spectra for the native luciferase and its mutant forms MT2, MT3, and MT4.
Spectra of bioluminescence for the initial luciferase and the MT8 mutant were identical at all studied pH values; hence the S118C replacement had no influence. The lmax value of bioluminescence for wild type luciferase shifted from 566 to 618 nm upon decrease in pH from 7.8 to 5.8 (Fig. 1a). Slight increase in the red component was observed for mutant MT2 upon lowering pH, and the contributions of green and red components became almost equal at pH 6.0 (Fig. 1b). Mutant MT2 contains replacements F16L and I19T. The mutation of the corresponding residue F14R in luciferase from P. pyralis strongly reduced the contribution of the red component in the bioluminescence spectrum at pH 6.5 [13]. The F16L replacement is apparently responsible for the change in bioluminescence spectra of mutant MT2. Residue I19 is far from the active center, and its replacement could hardly influence the bioluminescence spectrum MT2. The analysis of luciferase sequence alignments shows (Fig. 2) that the majority of firefly luciferases possessing pH-sensitive bioluminescence spectra have aromatic residues Phe and Tyr at position 16, while luciferases with pH-insensitive spectra have aliphatic Leu, Arg, and Lys.Fig. 1. Bioluminescence spectra of native luciferase (a) and mutants MT2 (b), MT3, and MT4 (c) at pH 7.8, 6.3, 6.1, and 5.8 (curves 1-4, respectively).
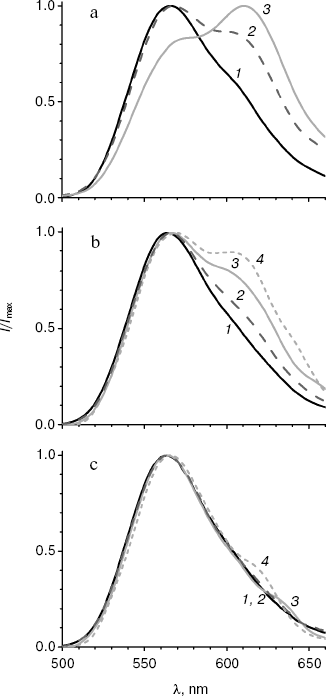
The analysis of possible conformations of residue L16 showed that the presence of a more flexible aliphatic instead of a rigid aromatic residue in this position can lead to more dense hydrophobic packing. This probably stabilizes the enzyme conformation, hindering its change on decreasing pH, which keeps mainly the green luminescence.Fig. 2. Amino acid sequence alignment for fragments of several luciferases in the regions 10-22 and 34-42 according to the L. mingrelica firefly luciferase numbering.
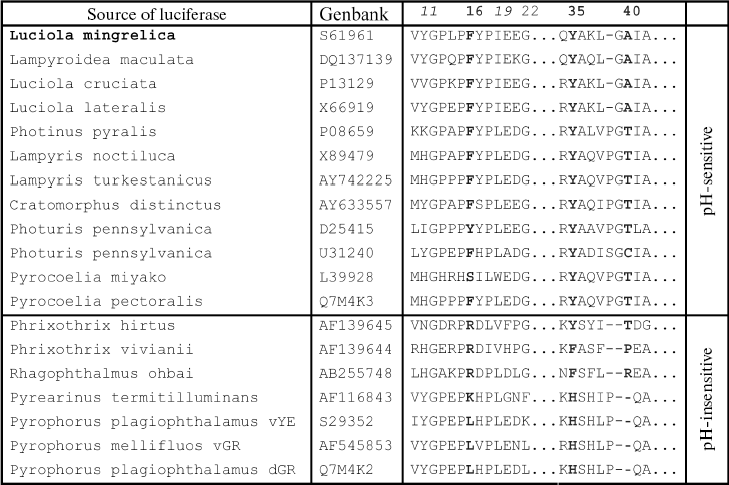
Bioluminescence spectra for mutants MT3, MT4, and MT6 were found to be identical at pH 7.8. At pH < 7.8 the MT6 spectrum was only slightly wider than that for mutant MT3. Spectra for mutants MT3 and MT4 were identical in the whole studied pH range (Fig. 1c) and were practically unchanged on decrease in pH. Mutants MT3 and MT4 had replacement of the same residue, Tyr35, to Asn and His, respectively. Hence, due to the replacement of this one residue (Y35) the bioluminescence spectrum of the mutant luciferase becomes virtually insensitive to pH. Also, mutant MT4 contains the replacement K191R. This residue is located outside the active center on the surface, and its backbone group is solvent-accessible; therefore, its replacement to the similar residue Arg should not influence the bioluminescence spectrum of MT4. The Y35N,H replacement leads to an effect similar to that obtained earlier for L. mingrelica luciferase H433Y substitution [28]. Luciferases containing His or Asn residues at position 433 emitted green bioluminescence at pH 7.8, and enzyme with Tyr emitted red light. Green luminescence was observed for mutants MT3 and MT4, containing Asn or His residues in position 35, at pH ~6.0, while the native luciferase with Tyr35 emitted red light.
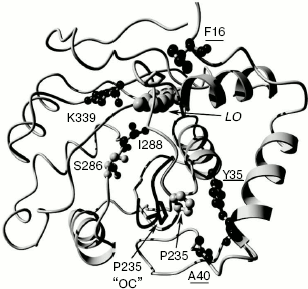
Mutant enzyme MT6 contains substitutions Y11F, F16L, A40S, and S118C, and also has a practically pH-insensitive spectrum of bioluminescence. As above, substitution S118C does not influence the bioluminescence spectrum (mutant MT8). Tyr11 is a surface residue far from the active center. A number of luciferases have V, N, I, K, and H residues instead (Fig. 2), but pH sensitivity of their bioluminescence spectra is preserved. So Tyr11 substitution with the similar residue Phe should not influence the bioluminescence spectrum. It shown above, the mutant MT2 containing substitution F16L has significantly reduced pH sensitivity of its bioluminescence spectrum. Therefore, the additional contribution of the A40S substitution led to a bioluminescence spectrum practically insensitive to pH. Residue A40 is located near residue Y35 (Fig. 3). It is probable that residue Ser40 in MT6 forms a hydrogen bond with a neighboring flexible chain resulting in the stabilization of the enzyme structure necessary for green bioluminescence.
Thus, the presented data show that an alteration of spectral properties of the mutants is likely to be caused only by the substitution of residues F16, Y35, and A40. These residues are neighboring to a loop forming one of the walls of the luciferin-binding channel (Fig. 3).Fig. 3. Model of the complex of firefly luciferase from L. mingrelica in “closed” conformation (E-DLSA). Residues whose mutations caused a decrease in pH sensitivity of their bioluminescence spectra are underlined. LO - oxyluciferin/luciferin position. The location of the loop region 233-237 in the case of the “open” conformation (“OC”) is shown in black. Molecular graphics were created with YASARA (www.yasara.org) and PovRay (www.povray.org).
It is known that the shift in bioluminescence spectra of firefly luciferases to the red region is observed not only on decreasing pH, but also on increase in temperature [1]. Both factors disrupt the rigid orientation of groups necessary for the formation of the green emitter, resulting in an increased contribution of red bioluminescence. To estimate the influence of temperature on bioluminescence spectra of the enzymes mutated at Y35, bioluminescence spectra were registered for MT3 and wild-type luciferase at 10, 20, 25, and 42°C.
The bioluminescence spectrum of MT3 is only a little wider at 42°C as that at 25°C (Fig. 4b), and it virtually coincides with the spectrum observed at 25°C for the wild-type luciferase. The spectrum of the latter has a plateau at 580-600 nm instead of a peak at 42°C, i.e. intensities of green and red emitters in the spectrum become equal. Thus, the bioluminescence spectrum of the Y35N mutant appeared to be not only more pH insensitive, but also thermostable. It should be noted that the wider spectrum of the native luciferase at 25°C (table) is also caused by the temperature effect: temperature decrease to 10°C decrease the half-width to 62 nm, i.e. only the green emitted is present (Fig. 4a).Fig. 4. Bioluminescence spectra of wild-type luciferase (a) and mutant enzyme MT3 (b) at 10, 20, 25, and 42°C (curves 1-4, respectively). Experiments were performed in pH 7.8 buffer.
In the literature, there is still no agreement about the chemical structure of green and red emitters of firefly luciferase. According to one conception, the red emitter is a ketonic form, and the green is an enolic or enolate form of oxyluciferin [12, 28]. According to another hypothesis, the emitter is an anionic ketoform of oxyluciferin, and such a significant shift of lmax is explained by a transition between two resonance forms releasing quanta of light with different energies [29]. However, there is no doubt about the conclusion that irrespective of the concrete molecular structure of the emitting forms, the structure of the protein environment of an emitter, the degree of its polarization, orientation, and flexibility of key amino acid groups contribute significantly to the spectral parameters of luciferase bioluminescence. Crystal structures of complexes of native and mutant (substitution S286N leading to red bioluminescence) luciferases from L. cruciata with an intermediate product analog (DLSA) (complexes E-DLSA and mE-DLSA) as well as a structure of a complex of the native luciferase with reaction product (oxyluciferin) and AMP (E-LO-AMP) were obtained earlier [22]. Based on analysis of the structures, the authors drew the conclusion that the active center in E-DLSA is in “closed” conformation. This leads both to rigid and mainly hydrophobic microenvironment of oxyluciferin and to the formation of the green emitter. Due to the change in orientation of I288, the mE-DLSA complex has an “open” conformation with less rigid environment of the substrate, so it is more accessible in the phenol ring region, and the relaxation of the excited molecule is possible to a form which will emit red bioluminescence upon transition to the ground state [22]. It should be noted that the spatial structures of these complexes almost coincide. They are notably different only in the orientation of residues S286 and I288, as well as in evident displacement of loop region 233-237 and at weak displacement of the loop 355-360 (Fig. 3). In the E-DLSA complex, residue P235 from loop 233-237 becomes close to residue Y35, being displaced by ~3.9 Å compared to complex E-LO-AMP. The displacement does not occur in the mE-DLSA complex, and it could be assumed that this displacement is necessary for the formation of the enzyme conformation resulting in green luminescence. Thus, residue Y35 is neighboring to the loop 233-237, whose position is important for the maintenance of a “closed” conformation of the luciferase active center leading to the green bioluminescence. It is quite probable that decrease in pH leads to similar “open”, less rigid conformation of the enzyme active center, as in the case of mutation S286N, causing the shift of the bioluminescence spectrum to the red region (Fig. 1a). When bulky aromatic residue Y35 was substituted with smaller residues Asn or His, dense packing near residues 35 and 225 becomes more stable, loop 233-237 preserves its position even at decreased pH, and therefore the “closed” conformation is not disrupted. A number of literature data also show the importance of this site for the formation of bioluminescence color. Thus, substitution of residue Val233 adjacent to Tyr35 by Asn in P. pyralis luciferase led to a mutant with red color [30], and its substitution with Ile in luciferase from L. cruciata led to a mutant with greener bioluminescence than that of the native enzyme [31]. Residue Tyr35 is conserved in all firefly luciferases. Click-beetle luciferases have His residue in this position, so it is probable that this is the one of positions contributing to pH insensitivity of their bioluminescence spectra. Ease in obtaining mutations that have decreased pH sensitivity of bioluminescence apparently shows that this property in fireflies is under the action of stabilizing selection. It should be noted that the formation of the microenvironment of oxyluciferin in the active center and the emission of green or red bioluminescence are defined by the balance of interactions of many amino acid residues in an extensive area of the enzyme. In particular, such factors as a network of hydrogen bonds formed by residues R220, N231, S286, E313, and K339 [32] in the region of luciferin-binding channel, hydrophobic packing of internal residues [14], interactions in the ATP-binding center [33] and between two flexible domains of luciferase [13, 28] play important roles. The conformation of loop 233-237 could be just one of the necessary factors. Therefore, there are no strictly specific residues explaining the dependence of bioluminescence spectra of luciferase on pH. A number of mutations far from each other that are directly or indirectly disrupting the necessary interactions and leading to red luminescence contribution [12, 28, 30], as well as mutations stabilizing the structure of the active center and lowering the dependence of the bioluminescence spectrum on external conditions [13, 14].
DNA sequencing was performed at the Inter-Institutional Center “GENOM”, Institute of Molecular Biology, Russian Academy of Sciences (http://www.genome-centre.narod.ru) organized with the support of the Russian Foundation for Basic Research (grant 00-04-55000).
This work was financially supported by the Russian Foundation for Basic Research (grant 08-04-00624).
REFERENCES
1.DeLuca, M. (1976) Adv. Enzymol., 44,
37-68.
2.Seliger, H. H., and McElroy, W. D. (1960) Arch.
Biochem. Biophys., 88, 136-141.
3.Ando, Y., Niwa, K., Yamada, N., Enomoto, T., Irie,
T., Kubota, H., Ohmiya, Y., and Akiyama, H. (2008) Nature
Photon., 2, 44-47.
4.Viviani, V. R., and Ohmiya, Y. (2006) in
Photoproteins in Bioanalysis (Daunert, S., and Deo, S. K., eds.)
Wiley-VCH, Weinheim, pp. 49-63.
5.Lundin, A. (2000) Meth. Enzymol.,
305, 346-370.
6.Viviani, V. R. (2002) Cell. Mol. Life
Sci., 59, 1833-1850.
7.Xinhua, F., Ohba, N., Vencl, F. V., and Chaoliang,
L. (2006) Can. Entomol., 138, 860-870.
8.Morton, R. A., Hopkins, T. A., and Seliger, H. H.
(1969) Biochemistry, 8, 1598-1607.
9.Liu, Y.-J., De Vico, L., and Lindh, R. (2008) J.
Photochem. Photobiol. A: Chem., 194, 261-267.
10.Nakatani, N., Hasegawa, J. Y., and Nakatsuji, H.
(2007) J. Am. Chem. Soc., 129, 8756-8765.
11.Viviani, V. R., Arnoldi, F. G. C., Ogawa, F. T.,
and Brochetto-Braga, M. (2007) Luminescence, 22,
362-369.
12.Ugarova, N. N., and Brovko, L. Y. (2002)
Luminescence, 17, 321-330.
13.Law, G. H., Gandelman, O. A., Tisi, L. C., Lowe,
C. R., and Murray, J. A. (2006) Biochem. J., 397,
305-312.
14.Kajiyama, N., and Nakano, E. (1991) Protein
Eng., 4, 691-693.
15.Talebarovskaya, I. K., Katkova, V. A., Rigova, V.
V., Shegolev, A. A., and Berezin, I. V. (1983) Author's Certificate No.
1192324.
16.Tu, Z., He, G., Li, K. X., Chen, M. J., Chang,
J., Chen, L., Yao, Q., Liu, D. P., Ye, H., Shi, J., and Wu, X. (2005)
Electron. J. Biotechn., 8, 114-120.
17.Lundovskich, I. A., Leontieva, O. V., Dementieva,
E. I., and Ugarova, N. N. (1999) in Bioluminescence and
Chemiluminescence: Perspectives for 21st Century (Roda, A.,
Pazzagli, M., Kricka, L. J., and Stanley, P. E., eds.) John Wiley &
Sons, Chichester, pp. 420-424.
18.Ho, S. N., Hunt, H. D., Horton, R. M., Pullen, J.
K., and Pease, L. R. (1989) Gene, 77, 51-59.
19.Cirino, P. C., Mayer, K. M., and Umeno, D. (2003)
Meth. Mol. Biol., 231, 3-9.
20.Wood, K. V., and DeLuca, M. (1987) Analyt.
Biochem., 161, 501-507.
21.Studier, F. W. (2005) Protein Expression and
Purification, 41, 207-234.
22.Nakatsu, T., Ichiyama, S., Hiratake, J.,
Saldanha, A., Kobashi, N., Sakata, K., and Kato, H. (2006)
Nature, 440, 372-376.
23.Berman, H. M., Westbrook, J., Feng, Z.,
Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., and Bourne,
P. E. (2000) Nucleic Acids Res., 28, 235-242.
24.Rodriguez, R., Chinea, G., Lopez, N., Pons, T.,
and Vriend, G. (1998) Bioinformatics, 14, 523-528.
25.Fiser, A., and Sali, A. (2003)
Bioinformatics, 19, 2500-2501.
26.Chenna, R., Sugawara, H., Koike, T., Lopez, R.,
Gibson, T. J., Higgins, D. G., and Thompson, J. D. (2003) Nucleic
Acids Res., 31, 3497-3500.
27.Shafikhani, S., Siegel, R. A., Ferrari, E., and
Schellenberger, V. (1997) Biotechniques, 23, 304-310.
28.Ugarova, N. N., Maloshenok, L. G., Uporov, I. V.,
and Koksharov, M. I. (2005) Biochemistry (Moscow), 70,
1262-1267.
29.Branchini, B. R., Southworth, T. L., Murtiashaw,
M. H., Magyar, R. A., Gonzalez, S. A., Ruggiero, M. C., and Stroh, J.
G. (2004) Biochemistry, 43, 7255-7262.
30.Branchini, B. R., Southworth, T. L., Khattak, N.
F., Murtiashaw, M. H., and Fleet, S. E. (2004) Proc. SPIE,
5329, 170-177.
31.Kajiyama, N., and Nakano, E. (1994) US Patent No.
5330906.
32.Viviani, V. R., Oehlmeyer, T. L., Arnoldi, F. G.,
and Brochetto-Braga, M. R. (2005) Photochem. Photobiol.,
81, 843-848.
33.Tisi, L. C., Law, G. H., Gandelman, O., Lowe, C.
R., and Murray, J. A. H. (2002) in Bioluminescence and
Chemiluminescence: Progress and Current Applications (Stanley, P.
E., and Kricka, L. J., eds.) World Scientific, Singapore, pp.
57-60.