Stage Specific Developmental Changes in the Mitochondrial and Surface Membrane Associated Redox Systems of Leishmania donovani Promastigote and Amastigote
B. Chakraborty, S. Biswas, S. Mondal, and T. Bera*
Division of Medicinal Biochemistry, Department of Pharmaceutical Technology, Jadavpur University, Kolkata 700032, West Bengal, India; fax: +91(033)2414-6677; E-mail: bidisha1219@yahoo.co.in; shibendu4132@gmail.com; subhasish19802002@yahoo.co.in; dr_tanmoybera@yahoo.co.in* To whom correspondence should be addressed.
Received March 2, 2009; Revision received October 19, 2009
Energy metabolism of Leishmania donovani parasite has been investigated under conditions imitating intralysosomal-like environment in the host organism. Trans-plasma membrane electron transport and oxygen uptake were inhibited progressively when promastigote cells were exposed to pH 5.5 and 37°C. A special feature of the respiratory chain in amastigote was the absence of complex I, II, and IV. When L. donovani was grown at pH 5.5 and 37°C, the acid excretory product succinate was increased in comparison to cells grown at pH 7.5 and 24°C. The findings of this study showed that the amastigote form catabolized fatty acid to excrete succinic acid when oxidative phosphorylation was impaired. Amastigote mitochondria failed to generate membrane potential by oxidizable substrates. On the other hand, the amastigote cell showed absorbance change of safranine O when fatty acid was the oxidizable substrate. The safranine signal was completely reversed by valinomycin, carbonyl cyanide 4-(trifluromethoxy)phenylhydrazone, malonate, and oxaloacetate. Our data suggest that the generation of metabolic energy from succinate/H+ efflux will contribute to energy requiring process of amastigote significantly. On the basis of these results, we conclude that due to absence of oxidative phosphorylation in amastigotes, energy linked functions in amastigotes might occur through fumarate reduction leading to ΔpH generation by succinate excretion.
KEY WORDS: Leishmania donovani promastigote and amastigote, mitochondria, differentiation, stage specificDOI: 10.1134/S0006297910040140
Abbreviations: ALA, α-lipoic acid; Am, amastigote; CoQ2H2, reduced coenzyme Q2; DCCD, dicyclohexylcarbodiimide; DCPIP, 2,6-dichlorophenolindophenol sodium salt; DPC, digitonin permeabilized cell; DTNS, 5,5′-dithiobis(2-nitroaniline-N-sulfonic acid); FCCP, carbonyl cyanide 4-(trifluromethoxy)phenylhydrazone; LDC, Leishmania donovani cell; NQSA, 1,2-naphthoquinone-4-sulfonic acid sodium salt; Pro, promastigote; RCR, respiratory control ratio; TMPD, N,N,N′,N′-tetramethyl-p-phenylenediamine; transPMET, trans-plasma membrane electron transport.
Leishmania donovani, the causative agent of human visceral
leishmaniasis, is a digenetic parasitic protozoon that cycles between
phagolysosomes of mammalian macrophages and the alimentary tract of
sand flies [1]. In the vector, they grow as
extracellular flagellated promastigotes, in the host they are
phagocytized by macrophages and proliferate as intracellular
non-flagellated amastigotes. Both forms are adapted to live in hostile
hydrolytic environments, and it is believed that specific molecules
expressed on the parasite cell surface membrane play a key protective
role [2].
During transition through these different extra- and intracellular environments, Leishmania is exposed to many changes in their living conditions. These changes include the elevated temperature of the mammalian host, toxic oxidants produced during phagocytosis by macrophages, acidic pH and proteases encountered in the macrophage phagolysosome, variations in the availability and types of nutrients, as well as the availability of oxygen. The mechanism that allows parasites to withstand these noxious stimuli is probably critical for their survival [3].
The drugs for the treatment of visceral leishmaniasis are pentavalent antimony (SbV)-containing drug, amphotericin B, and chalcone derivatives. Evidences showed that the intracellular amastigote form of Leishmania was more susceptible than promastigote to pentavalent antimony-containing drugs [4], amphotericin B [5], and chalcone derivatives [6]. Differential sensitivities of these drugs with respect to amastigotes and host cells clearly indicate the presence of distinguishable cell biological and biochemical processes in the amastigote. Therefore, characterization of stage-specific metabolic steps is an important step in development of new therapeutic agents against the intracellular stage, which is responsible for the maintenance of leishmaniasis in mammals.
Studies during the last decade indicated that shifting promastigotes to an intralysosomal-like environment (e.g. 37°C and pH 5.5 at 5% CO2 environment for visceral species) induced differentiation into amastigotes in host-free culture [7, 8]. Acidic pH and heat induced growth arrest in promastigotes route the heat-adapted promastigotes to differentiate into amastigotes [9]. Such an experimental system has already been used by several laboratories to investigate various stage-regulated functions in L. donovani [4, 10] and has also been developed in our laboratory to investigate various stage-regulated functions in L. donovani. Characterization of axenic amastigotes of the Leishmania species has demonstrated that they resemble animal-derived amastigotes [8]. Axenic amastigotes are virulent as they infect hamsters and macrophage cell lines. Moreover, differentiation in host-free culture resumes virulence of long-term attenuated promastigotes [8].
The leishmaniae and other trypanosomatids are unique in that they contain a single mitochondrion per cell which convolutes and ramifies in situ and occupies around 12% of the cell volume, and it is associated with the kinetoplast to form a kinetoplast–mitochondrial complex [11]. Quantitative morphometry of electron micrographs of Leishmania spp. demonstrated that after 3 h of heat treatment there was no change in mitochondrial morphology, but after 6 h of heat treatment the mitochondria have lost their cristae and no longer possess a clearly defined mitochondrial double membrane [12]. Studies on these aspects have been greatly hampered by difficulties in isolation of intact mitochondria from trypanosomatids. Isolation of this organelle is difficult because of the large size and ramification of the single mitochondrion and the presence of microtubule arrays adhering to the inner side of the cell membrane of the trypanosomatids [13]. As a consequence, greater force is required to break the cells than the force necessary to disrupt the mitochondrial membrane [13, 14]. To overcome these difficulties, digitonized cells have been useful as an experimental model.
Martin and Mukkada [14] reported evidence for the presence of complex I, II, III, and IV in the respiratory chain of L. tropica promastigote. In contrast, Santhamma and Bhaduri claimed [15] that complex I is absent from the respiratory chain of L. donovani promastigote. No data are available on electron transport chain, energy coupling, and oxidative phosphorylation in the amastigote form of Leishmania.
Recent reports have indicated that the plasma membrane of L. donovani promastigotes can carry out plasma membrane electron transport [16, 17]. The finding that heat transformed, acidic pH stabilized L. donovani cells down-regulate plasma membrane and mitochondrial electron transport as well as oxygen uptake prompted us to investigate the nature of energy metabolism in L. donovani amastigotes. Many lower eukaryotes can survive hypoxic or anaerobic conditions via a fermentative pathway that involves the use of the reduction of endogenously produced fumarate as an electron sink. They are highly adapted for prolonged survival or even continuous functioning in the absence of oxygen, whereas many of them are adapted to alternating periods in the presence and absence of oxygen [18]. Some anaerobically functioning eukaryotes, such as yeast and certain fishes, can survive without mitochondrial energy metabolism via cytosolic fermentations in which the NADH produced during glycolysis is consumed during the reduction of pyruvate to lactate or fumarate to succinate, which are subsequently excreted as end-products [19]. The present communication deals with the differentiation of L. donovani promastigote and amastigote electron transport chains and the developmental changes. This study was also undertaken to elucidate whether the fumarate reduction leading to succinate excretion is a fundamental process to generate proton motive force that might be a drug target [20].
MATERIALS AND METHODS
Materials. All chemicals unless otherwise mentioned were from Sigma (USA). 5,5′-Dithiobis(2-nitroaniline-N-sulfonic acid) (DTNS) was synthesized in our laboratory [16].
Promastigote culture method. Leishmania donovani promastigote clonal strain MHOM/IN/1978/UR6, a clinical isolate from a confirmed Indian kala-azar patient [21], was grown at 24°C on blood agar medium, pH 7.5 [22]. Cells were harvested after 3 days growth in late log phase by centrifugation at 500g for 10 min at 4°C twice in cold Tris-sucrose-salt solution (250 mM sucrose, 50 mM NaCl, 20 mM KCl, 1 mM EDTA, 20 mM Tris, pH 7.2). Viability of the harvested cells was monitored microscopically by the trypan blue exclusion method [23].
Generation of axenic amastigotes. Leishmania donovani amastigote strain MHOM/IN/1978/UR6 were grown and maintained as described by Debrabant et al. [8]. Axenically grown amastigotes of L. donovani were maintained at 37°C with 5% CO2 by weekly sub-passages in MAA/20 (medium for axenically grown amastigotes) at pH 5.5 in 177 cm2 Petri dishes [24]. Under these conditions, promastigotes differentiated to amastigotes within 120 h. Cultures were maintained by 1 : 3 dilutions once in a week. The axenic amastigotes remained stable in culture for a long time. Axenic amastigotes were routinely recycled every 10 weeks by differentiation back to promastigotes, and in parallel, initiating a new line of promastigotes. Amastigotes were transformed to promastigotes by centrifugation of amastigotes (1000g at room temperature for 10 min), suspension in promastigote medium [25], and incubation at 24°C. Under these conditions amastigotes differentiated to promastigotes within 72 h. In all of the experiments in this paper where established amastigotes were required, only 4-8-week-old amastigotes were used.
Cell fractionation and isolation of kinetoplast mitochondria. Mitochondria were isolated under hypotonic conditions by a modification of the method as described by Hauser et al. [26]. Kinetoplast mitochondria were prepared from L. donovani promastigotes in late logarithmic phase of growth. All operations were performed at 4°C. The cells were washed twice with buffer A (140 mM NaCl, 20 mM KCl, 20 mM Tris, 1 mM EDTA, pH 7.5) and suspended in hypo-osmotic buffer (5 mM Hepes, pH 7.7, 1 mM EDTA) to a concentration of approximately 5 mg cells/ml. Clumping of cells was prevented by agitating the cell suspension with a magnetic stirrer. After 15 min of swelling, the cell suspension was homogenized in a Potter–Elvehjem homogenizer with a Teflon pestle at 2000 rpm. This preparation was monitored microscopically for cell breakage. The resulting homogenate, adjusted to pH 7.5 with 1 N KOH, was centrifuged at 700g for 7 min. The supernatant was centrifuged again as above. The precipitates from the two centrifugations were discarded, and supernatants were centrifuged at 8000g for 15 min. The fraction rich in nuclei, flagella, and large cell fragments was collected in the 700g pellet and mitochondrial fraction in the 8000g pellet. After removing the supernatant by aspiration, each pellet was resuspended in DNase buffer (20 mM Mops-NaOH, 350 mM sucrose, 0.3% bovine serum albumin (BSA), 5 mM magnesium acetate, 1 mM EGTA, 100 µg/ml DNase I, pH 7.7) for 30 min. After DNase I digestion, the lysate was loaded on 20-35% (w/v) Percoll gradients. The final fraction of mitochondria was resuspended at high protein concentration (30-50 mg/ml) in isolation buffer (20 mM Mops-NaOH, 350 mM sucrose, 20 mg/ml fatty acid and protease-free BSA, 20 mM potassium acetate, 5 mM magnesium acetate, and 1 mM EGTA, pH 7.0) with 0.15 mg/ml soybean trypsin inhibitor and 0.02 mg/ml leupeptin.
Amastigote mitochondria were isolated under isotonic condition by modifying the procedure described by Hauser et al [26]. Cells were lysed by intracytoplasmic cavitation with nitrogen gas in isolation buffer at a concentration of 5 mg cells/ml. Two subsequent low speed spins (500g) were performed to remove the remaining intact cells. After a DNase I digestion, the lysate was applied to 20-35% (w/v) Percoll gradients. The final fraction of mitochondria was resuspended at high protein concentration (30-50 mg/ml) in isolation buffer containing 0.15 mg/ml soybean trypsin inhibitor and 0.02 mg/ml leupeptin.
Mitochondria and submitochondrial particles were also prepared from L. donovani promastigote, amastigote, and rat liver according to an established method [27].
Digitonin permeabilization of cell. Leishmania donovani promastigote and/or amastigote cells were collected, washed once by buffer A, and resuspended in isolation buffer (20 mM Mops-NaOH, 0.3% BSA, 350 mM sucrose, 20 mM potassium acetate, 5 mM magnesium acetate, and 1 mM EGTA, pH 7.0). Cells were permeabilized with various amounts of digitonin (25-300 µg/mg protein) and incubated on ice for 10 min. After incubation, the cells were centrifuged at 6000g for 7 min. The enzymatic activities that were released from the cells were assayed in the supernatant. Pellets were resuspended in assay buffer.
Respiratory measurements. Rates of oxygen consumption were measured in buffer B (100 mM Mops-NaOH, 20 mg/ml BSA, 300 mM sucrose, 7 mM dipotassium hydrogen phosphate, 5 mM magnesium acetate, and 1 mM EGTA, pH 7.0) at 25°C in a water-jacketed DW1 Hansatech oxygraph with 1-ml glass chamber (Hansatech Instruments Ltd., UK) containing a Clark type polarographic oxygen electrode. The solubility of oxygen in air-saturated temperature-equilibrated medium was taken to be 480 ng-atoms/ml at 25°C and 760 mm Hg [28].
Determination of enzyme activity. Measurement of ferricyanide reduction by L. donovani cells. Ferrocyanide was quantified by a published procedure [16, 17] using 1,10-phenanthroline complex as described by Avron and Shavit [29]. The incubation was carried out at 24°C for L. donovani promastigotes and at 37°C for L. donovani amastigotes.
α-Lipoic acid (ALA), DTNS, and 1,2-naphthoquinone-4-sulfonic acid (NQSA) reduction assay. ALA, DTNS, and NQSA reduction by L. donovani promastigote cells were assayed as described earlier [16] as the formation of ferrocyanide as a result of the reduction of ferricyanide by dihydrolipoic acid, 5-mercapto-2-nitroaniline-N-sulfonic acid, and hydroquinone of NQSA. Ferrocyanide was estimated according to the method of Avron and Shavit [29]. The reaction mixtures were incubated for 10 min at 24°C for L. donovani promastigotes and at 37°C for L. donovani amastigotes.
Measurement of 2,6-dichlorophenolindophenol sodium salt (DCPIP) reduction by L. donovani cells. Leishmania donovani cells (LDC) (5 mg protein/ml) were incubated at 24°C for L. donovani promastigotes and at 37°C for L. donovani amastigotes in phosphate buffered saline (PBS), pH 7.0, along with 5 mM D-glucose. The reaction was started by the addition of 60 µM DCPIP. The rate of DCPIP reduction was followed by the decrease in absorbance at 600 nm during 1 min in a double beam Shimadzu UV-visible spectrophotometer, model UV-2450. The control cuvette contained either L. donovani promastigotes or L. donovani amastigotes in phosphate buffered saline (PBS), pH 7.0, along with 5 mM D-glucose minus DCPIP and was monitored simultaneously. The extinction coefficient for DCPIP at 600 nm was taken as 21.2 mM–1·cm–1 at pH 7.0.
NADH:duroquinone oxidoreductase activity. This enzyme activity was determined using 0.06 mg/ml of mitochondrial protein in 20 mM K2HPO4, 2 mM KCN, 0.1 mM EDTA, 0.3 mM duroquinone, 0.13 mM NADH, pH 7.2 [30, 31]. The decrease in absorbance at 340 nm was recorded. Rotenone at 30 µM was used as a specific inhibitor of complex I.
Succinate dehydrogenase activity. Succinate dehydrogenase activity was measured spectrophotometrically at 600 nm (ε = 21.2 mM–1·cm–1) using enzyme preparation from either promastigote or amastigote, 5 mM succinate, 60 µM DCPIP in assay buffer (20 mM Mops, 200 mM sucrose, 70 mM potassium acetate, 1 mM EGTA, pH 7.0) with 5 mM magnesium acetate. The enzymatic reaction was carried out in 1 ml reaction volume.
CoQ2H2-cytochrome c oxidoreductase activity. Reduced coenzyme Q2 (CoQ2H2) was prepared according to the method of Rieske [32]. CoQ2H2-cytochrome c oxidoreductase activity was determined at 550 nm (ε = 18.9 mM–1·cm–1) using enzyme preparation from either promastigote or amastigote, 20 µM cytochrome c, and 30 µM CoQ2H2 with 5 mM magnesium acetate in assay buffer, pH 7.0. For promastigote, 2 mM KCN was added in the reaction mixture. For amastigote, the reaction mixture was bubbled with oxygen-free N2 for 2 min before the enzyme was added and kept anaerobic by adding silicone oil at the top of the cuvette. The reaction was initiated by the addition of 20 µM cytochrome c through the silicone oil.
Identification and quantification of acid end product. Succinate in spent medium derived from stationary-phase cultures of L. donovani promastigotes and amastigotes was identified and quantified using the procedure of Bril [33] and finally by HPLC. The spent medium was deproteinized by adding 1/5 of its volume of 5% metaphosphoric acid solution and centrifugation. The protein-free medium was extracted with diethyl ether. The ether was evaporated under vacuum. The residue was dissolved in HPLC grade water. Samples were filtered through 0.2 µm pore membrane filter (Sartorius Biotech, Germany). The injection and elution were carried out with a Rheodyne injector (Rohnert Park, USA) with a 20 µl loop and a pump (model 1150; GBC, Australia). The column (Supelcogel C-610H, 250 × 7.8 mm) and the pre-column (Supelcogel C-610H, 50 × 7.8 mm) were purchased from Supelco (Bellefonte, USA) and maintained at 40°C. The flow rate was 0.4 ml/min and the solvent system was H3PO4–H2O (1 : 99 v/v). For detection, the HPLC system consisted of a variable UV detector (model 480C, set at 210 nm) (Instrumentos CG, Brazil) and a data-handling system CBM-101 (Shimadzu, Japan). Analytical grade succinic acid purchased from Sigma was used as standard. To produce calibration curves, aliquots (7, 5, 2.5, and 1 ml) of the stock solution (10 mg/7 ml), corresponding to 10, 7.14, 3.57, and 1.42 mg, respectively, were diluted to 10 ml with HPLC grade water and 20 µl of each of these solutions corresponding to 20, 14.28, 7.14, and 2.84 µg were analyzed by HPLC. The calibration curves (quantity injected versus area under the peak) and the r value (correlation coefficient) were generated by the software for the calibration curve.
Estimation of mitochondrial membrane potential. Mitochondrial membrane potential of L. donovani promastigotes and amastigotes in situ were estimated spectrophotometrically using the indicating dye safranine O at 28°C [34]. Cells were incubated in medium containing 200 mM sucrose, 70 mM KCl, 10 mM Hepes, pH 7.0, 1 mM Mg-acetate, 0.4 mM EGTA, and digitonin (50 µg/mg LDC). Absorbance changes were monitored by a diode array spectrophotometer at the wavelength pair 500-533 nm. Each experiment was repeated at least three times.
Measurement of spectral changes in safranine O in the presence of L. donovani promastigote or amastigote cells. Safranine O uptake in L. donovani promastigotes and amastigotes were measured according to the procedure described by Åkerman and Jarvisalo [35] for hepatocyte cells. Promastigote cells were suspended at a final concentration of 2.5 mg/ml in buffer containing 140 mM NaCl, 5 mM Mg-acetate, 0.4 mM EGTA, 10 mM Hepes, pH 7.0, along with oxidizable substrates and 10 µg/ml oligomycin for 10 min before addition of 20 µM safranine O. Amastigote cells were suspended at a final concentration of 2.5 mg/ml in buffer containing 130 mM KCl, 10 mM Na-acetate, pH 4.5, 5 mM Mg-acetate, 0.4 mM EGTA along with 0.5 mM palmitate, 10 mM sodium bicarbonate, and 10 µg/ml oligomycin for 10 min before addition of 20 µM safranine O. The addition of 10 mM sodium bicarbonate to buffer of pH 4.5 resulted in final pH of buffer of 5.5. Observations were made at the wavelength pair 525-485 nm. Each experiment was repeated at least three times.
Protein estimation. LDC protein was determined by the biuret method in the presence of 0.2% deoxycholate [36]. One milligram of promastigote protein corresponds to 1.75·108 cells and 1 mg of amastigote protein corresponds to 1.14·108 cells. Cytosolic, mitochondrial, and plasma membrane proteins were determined by a modified Lowry method [37].
Statistical analysis. All experiments were performed in triplicate, with similar results obtained in at least three separate experiments. Statistical significance was determined by Student’s t-test. Significance was considered as P < 0.05.
RESULTS AND DISCUSSION
Respiratory properties of L. donovani cells during promastigote to amastigote differentiation. Upon transient exposure of L. donovani promastigote cells to pH 5.5 and 37°C (conditions which mimic intracellular growth of Leishmania), the rate of glucose stimulated oxygen uptake of cells progressively decreased with increase of incubation time (Figs. 1 and 2). Figure 1 represents the effect of pH and heat shock on the reversal of O2 uptake during incubation at pH 7.5 and 24°C. Complete regain of O2 uptake for 23-min pH- and heat-shocked promastigote was achieved after 48 h, whereas 180-min pH- and heat-shocked promastigote showed complete O2 uptake regain after 18 days. From the observations (Figs. 1 and 2) we can conclude that prolonged pH and heat shock on promastigotes required longer incubation at pH 7.5 and 24°C for complete recovery of O2 uptake. Amastigote cells derived from a 1-month-old cell line also showed very little endogenous O2 uptake (Table 1).
Fig. 1. Reversibility of down-regulated oxygen uptake in pH- and heat-shocked L. donovani promastigotes. Promastigote cells were incubated at pH 5.5 and 37°C in MAA/20 medium with 5% CO2 for 23 (1, open circles), 45 (2, closed circles), 90 (3, open squares), and 180 min (4, closed squares) followed by removal of cells by centrifugation. These cells were again incubated in promastigote liquid media at pH 7.5 and 24°C. Cells were removed at different time intervals from the medium by centrifugation. Oxygen uptake of pH- and heat-shocked and shock-recovered cells was measured in the presence of 5 mM D-glucose as described in “Materials and Methods”. Each point represents the average value of three experiments.
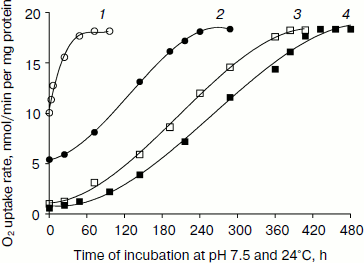
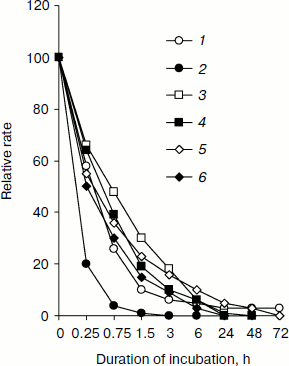
Table 1. Relative rate of substrate oxidation by mitochondria in digitonin-permeabilized L. donovani promastigotes and amastigotesFig. 2. Effect of duration of incubation at pH 5.5 and 37°C on oxygen uptake and trans-plasma membrane electron transport activities of L. donovani promastigote cells. Cells were incubated at 5% CO2 + 95% air in MAA/20 medium. Relative rate 100 was considered for DCPIP, ALA, DTNS, NQSA, and K3Fe(CN)6 as well as for oxygen uptake by L. donovani promastigote cells to be 19.32, 1.91, 0.66, 1.11, and 0.42 nmol/min per mg protein and 15.6 nmol O2/min per mg protein, respectively. Curves: 1) oxygen uptake; 2) DCPIP reduction; 3) ALA reduction; 4) DTNS reduction; 5) NQSA reduction; 6) ferricyanide reduction. Each point represents the average value of three experiments.
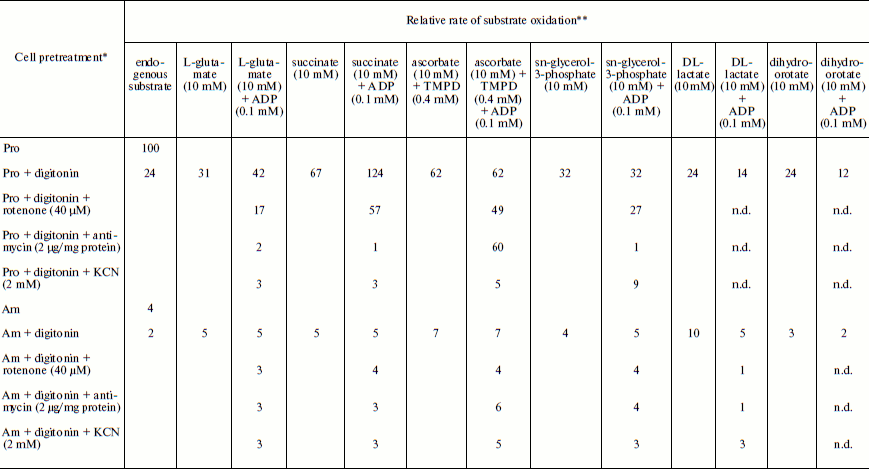
Note: n.d., not determined.
* Leishmania donovani promastigote and/or amastigote cells were treated,
where indicated, with 30 µg digitonin/mg cell protein for 3 min
before the addition of oxidizable substrates and/or electron transport inhibitor.
** Relative rate 100 was considered for L. donovani promastigote endogenous
oxygen uptake rate to be 15 nmol O2/min per mg cell protein.
Amastigote cells were taken from a cell line that had been subcultured 5 times at
7-day interval. Relative rate values represent the average of three experiments.
Figure 2 illustrates change in trans-plasma membrane electron transport (transPMET) activity of L. donovani promastigotes as a function of time after the cells were exposed to differentiation signal (pH 5.5 and 37°C). Evidence to support the existence of a trans-plasma membrane redox system in L. donovani promastigote arises from the reduction of the four non-permeable electron acceptors ALA, DTNS, NQSA, and ferricyanide by whole cells [16, 38, 39]. The transPMET activity as judged by the rate of reduction of non-permeable electron acceptors (ALA, DTNS, NQSA, ferricyanide, and DCPIP) and oxygen uptake were progressively diminished with increase of incubation time. Maximum fall of transPMET was observed with DCPIP followed by oxygen uptake and other non-permeable electron acceptors. Complete loss of transPMET activity was observed after 72 h of incubation. Only 4% residual oxygen uptake was retained even after 72 h of incubation. Incubation of promastigotes to pH 5.5 and 37°C induced biochemical differentiation. No change of morphology was observed during the first hour. Between 2 to 4 h cells aggregated and started to round, and almost 50% of the cell population lost their flagella. After 6 h 75% of the parasites rounded and lost their flagella, a process that completed at 24 h. Transferring axenic amastigotes to pH 7.5 and 24°C induced differentiation back to promastigotes as depicted in Fig. 1.
Respiratory activities of mitochondria in digitonin-permeabilized cell of L. donovani promastigotes and amastigotes and isolated mitochondria of liver and L. donovani promastigotes and amastigotes. Data reported in Table 1 demonstrate that L. donovani promastigotes and amastigotes could be selectively permeabilized by using digitonin at 30 µg/mg cell protein. Very little stimulation of oxygen consumption was achieved by the addition of NADH-dependent substrate glutamate to digitonin-permeabilized promastigotes in the presence of ADP (Table 1). Phosphorylation efficiency of complex I in promastigote appears to be 4 times less compared to complex I of liver mitochondria (Table 2). Even at very high concentration of rotenone, glutamate stimulated oxygen uptake was not completely inhibited (Table 1). Thus, results of Tables 1 and 2 suggest partial activity of complex I. Addition of antimycin A or cyanide completely blocked glutamate-stimulated oxygen uptake in digitonin permeabilized promastigotes (Table 1). These inhibitors suggest the presence of complexes III and IV in the pathway of electron transport from glutamate to oxygen. Malate/glutamate, a combination of substrates commonly used to demonstrate the presence of NADH-ubiquinone oxidoreductase (complex I) activity in mitochondrial preparations, was also unable to stimulate ADP phosphorylation in isolated promastigote mitochondria (Table 2). In contrast, under the same conditions isolated liver mitochondria showed profound stimulation in the presence of ADP. Other potential NADH-dependent substrates, such as 3-oxoglutarate, lactate, and pyruvate, were also unable to stimulate ADP phosphorylation in isolated promastigote mitochondria (Table 2). In addition, potential flavoprotein-linked substrates like sn-glycerol-3-phosphate and dihydroorotate failed to stimulate phosphorylation in digitonin permeabilized promastigotes (Table 1).
Table 2. Substrate oxidation by isolated
L. donovani promastigotes and amastigotes and liver
mitochondria*
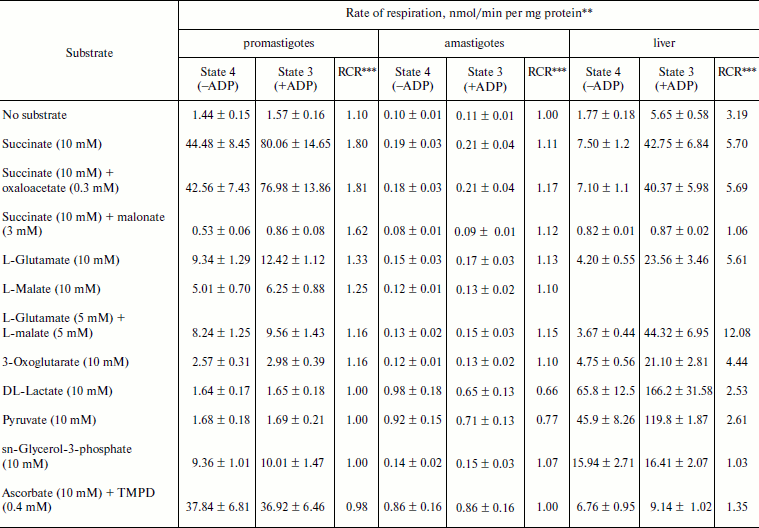
* L. donovani promastigotes and amastigotes and liver
mitochondria were isolated as described in “Materials and
Methods”. Concentrations of mitochondrial protein for
promastigotes, amastigotes, and liver cells were 2, 10, and
5 mg/ml, respectively.
** Rate values represent mean ± SD values of three experiments.
Oxygen uptake was recorded polarographically as described in
“Materials and Methods”.
*** RCR, respiratory control ratio.
The inclusion of the respiratory substrate system N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD)/ascorbate, which reduces respiratory complex IV, reinitiated respiration in digitonin permeabilized promastigotes and isolated promastigote mitochondria. Addition of ADP to this preparation resulted in no stimulation of respiration. Inclusion of cyanide into this system resulted in complete inhibition. Thus we can conclude that site IV of the mitochondrial electron transport chain of promastigotes is non-phosphorylating. Partial inhibition (21%) of TMPD/ascorbate-stimulated respiration in digitonin-permeabilized promastigote by rotenone suggests an alternative site of the cytochrome c-dependent electron transport pathway in promastigotes. Antimycin A had no effect on TMPD/ascorbate stimulated respiration in digitonin-permeabilized promastigotes (Table 1).
The failure of NADH-dependent substrates and TMPD/ascorbate to support increase of sufficient oxygen consumption in the presence of ADP in digitonin permeabilized promastigotes and isolated promastigote mitochondria suggests that the NADH dehydrogenase-ubiquinone (complex I) and cytochrome c oxidase segment (complex IV) are not functioning properly in L. donovani promastigotes. Addition of succinate to digitonin-permeabilized cells and isolated mitochondria of promastigotes resulted in profound stimulation of oxygen uptake. The subsequent addition of ADP induced the transition from resting (State 4) to phosphorylating (State 3) respiration. A respiratory control (State 3/State 4) of 1.8 was estimated in both digitonin-permeabilized promastigotes and isolated promastigote mitochondria (Tables 1 and 2). Addition of rotenone at 40 µM concentration partially inhibited (54%) State 3 respiration of succinate in digitonin-permeabilized promastigotes, which was then completely inhibited by antimycin A and cyanide (Table 1). Partial inhibition of succinate respiration by rotenone in digitonin-permeabilized promastigotes suggests the presence of an alternative succinate oxidation pathway in L. donovani promastigotes (Table 1).
Experiments using intact amastigote cells indicated loss of 96% of oxygen uptake when compared with the oxygen uptake of promastigote cells (Table 1). The addition of 30 µg digitonin/mg amastigote protein to a suspension of amastigotes incubated in buffer B containing flavoprotein-linked substrates such as succinate or sn-glycerol-3-phosphate or dihydroorotate was not able to stimulate respiration, whereas addition of DL-lactate resulted in moderate oxygen uptake stimulation in amastigotes (Table 1). The inability of L. donovani amastigote mitochondria to carry out energy linked functions such as respiration coupled with phosphorylation suggests that the amastigote form of Leishmania is less dependent on respiratory energy (Table 1). This might be the reason for the survival of amastigote cells within the phagolysosomes where apparent hypoxic conditions persist [40, 41]. Studies from Table 1 revealed that amastigotes were devoid of complex IV activity and failed to consume oxygen. Assays of complex I, II, and III suggest the presence of complex III in both promastigotes and amastigotes, whereas amastigotes failed to retain the activities of complex I and II (Table 3).
Table 3. Characterization of
NADH-duroquinone, succinate-DCPIP, and
CoQ2H2-cytochrome c oxidoreductase in
L. donovani promastigote and amastigote mitochondria and
submitochondrial particles
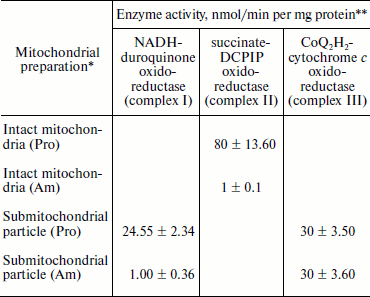
* Mitochondria and submitochondrial particles were prepared as described
in “Materials and Methods”.
** NADH-duroquinone, succinate-DCPIP, and
CoQ2H2-cytochrome c oxidoreductase
activities were assayed according to the procedures as given in
“Materials and Methods”. Rates were calculated from slopes
of linear absorbance changes. The values represent the average of three
experiments.
Effect of differentiation signal on the production of acid end product. Anaerobic metabolism of amastigotes suggested that fumarate might serve as electron sink of the reducing equivalents liberated during metabolism. This was evident from the amount of succinate formed during the growth of amastigotes (Table 4).
Table 4. Concentration of succinate in spent
medium obtained from stationary-phase cultures of L. donovani
promastigotes and amastigotes*

* Recovery of succinate from the spent medium of L. donovani
promastigotes and amastigotes is described in “Materials and
Methods”.
** Succinate was quantified by HPLC according to the procedure described
in “Materials and Methods”. The values represent average of
three experiments ± SD.
Safranine O uptake by promastigote and amastigote mitochondria and cells energized by respiratory substrates. Table 5 represents the rate of spectral shift change of safranine O due to the presence of oxidizable substrates in normal and digitonin-permeabilized promastigote and amastigote cells. In the absence of electron transport, mitochondrial energization can be supported by reverse operation of the F1F0-ATPase if ATP is available. Since the promastigote respiration is insensitive to rotenone [15], de-energization of these mitochondria was achieved by the addition of oligomycin and re-energization by the addition of oxidizable substrates. In the measurements using safranine O, uptake was maximally supported in promastigote and amastigote mitochondria by succinate and palmitate, respectively. Energization in promastigote and amastigote whole cells was maximally supported by D-glucose and palmitate in the presence of bicarbonate, respectively. Figure 3 shows the spectral responses of safranine O in amastigote cells energized by palmitate plus bicarbonate and their de-energization by FCCP (carbonyl cyanide 4-(trifluromethoxy)phenylhydrazone), valinomycin, malonate, and oxaloacetate.
Table 5. Comparison of the uptake of
safranine O in L. donovani promastigote and amastigote
mitochondria and cells by oxidizable substrates
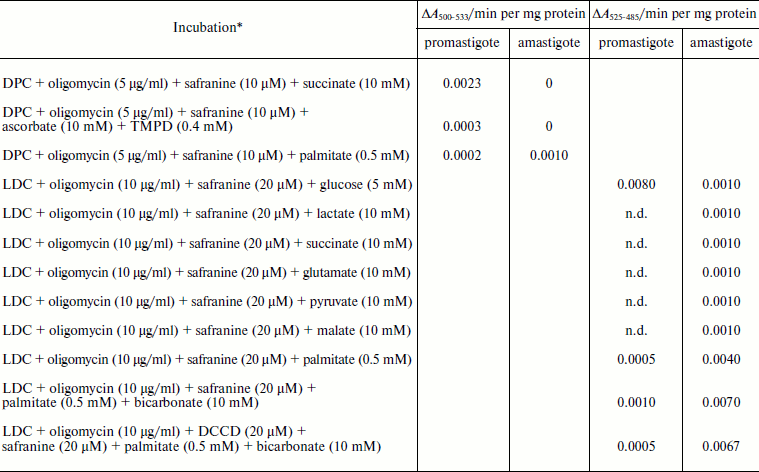
Note: DPC, digitonin-permeabilized cell; LDC, Leishmania donovani cell;
n.d., not determined.
* Safranine O uptake in digitonin permeabilized cell and normal cell was
carried out as described in “Materials and Methods”.
Oxidizable substrates were preincubated with LDC for 10 min before
addition of safranine O.
The safranine method has previously been used for determination of membrane potentials in bacterial vesicles [42] and mitochondria [43] as well as in Ehrlich ascites tumor cells [44] indicating that the spectrum of safranine in cells with de-energized mitochondria differs from that in the external medium. A CO2 requirement for amastigote growth (Table 5) often reflects the involvement of phosphoenolpyruvate carboxylase or pyruvate carboxylase in the synthesis of succinate. Martin et al. [45] demonstrated that in amastigotes pyruvate carboxylase is the principal enzyme catalyzing heterotrophic CO2 fixation to produce oxaloacetate from phosphoenolpyruvate. Evidence also suggests that the activities of these enzymes are comparatively higher in the glycosomes of amastigotes [46]. Such a pathway can provide dicarboxylic acids for biosynthetic processes and also contributes to NAD+ recycling and maintenance of the proton-motive force. Addition of protonophore FCCP with an external pH of 5.5 resulted in influx of H+ and efflux of safranine O, which caused a decrease in the absorbance of the dye at the respective wavelength pair (Fig. 3). Valinomycin in the presence of 130 mM K+ resulted in influx of K+ in exchange with efflux of cationic dye safranine O. Receptor-mediated efflux of succinate was antagonized by malonate and oxaloacetate, which resulted in de-energization and efflux of safranine O. Generation of membrane potential as judged by safranine uptake in amastigote cells was not inhibited by the ATPase inhibitor dicyclohexylcarbodiimide (DCCD), and hence proton-translocating ATPase was not involved in generation of membrane potential (Table 5). The same observation was observed in the amastigote form of MHOM/IN/1983/AG83 strain of Leishmania (data not presented).Fig. 3. Composite of records of the changes of safranine absorbance (525-485 nm) induced by its penetration into L. donovani amastigote suspension in response to energization by palmitate and bicarbonate followed by their de-energization as described in “Materials and Methods”. Incubation medium (1 ml) containing 2.5 mg amastigote cells, 10 µg/ml oligomycin, 0.5 mM palmitate, and 10 mM potassium bicarbonate was preincubated for 10 min before addition of safranine O. FCCP, malonate, oxaloacetate, and valinomycin were added to cause de-energization where indicated.
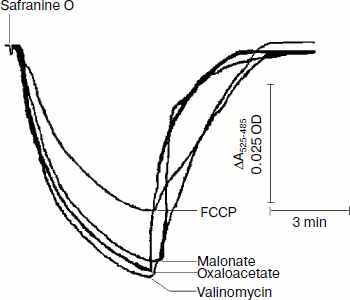
The life cycle of Leishmania is highly complex; the parasites at every step are morphologically and metabolically well adapted to a distinct compartment of their specific hosts. The recent development of a host-free differentiation system for Leishmania has enabled better insight into the early differentiation processes in this parasite. The major observations were: (i) temperature and pH shifts triggered oxygen uptake inhibition by as much as 96%, (ii) succinate failed to donate electrons in amastigote mitochondria, whereas DL-lactate was capable of donating electrons, (iii) amastigote mitochondria failed to display oxidative phosphorylation, (iv) excretion of protons coupled with succinate was a mechanism for the generation of a proton-motive force in amastigotes.
Amastigote cells continuously excrete relatively large quantities of succinic acid and other organic acids into their environment [46]. Michels et al. [20] in their “energy recycling model” proposed that electrogenic efflux of these organic end products via ion symport systems can lead to the generation of an electrochemical ion gradient across the cytoplasmic membrane, thus providing metabolic energy to the cell. Amastigote cells might be able to utilize succinate excretion as a metabolic energy source. It appears that dysfunctional mitochondria of amastigotes cause these cells to produce an inordinate amount of succinic acid (Table 4). An analogous observation for cancer cells is the inactive cytochrome c oxidase that leads to preferential utilization of glycolysis over aerobic respiration to produce ATP [47]. Leishmania amastigotes have been shown to contain greater activities than promastigotes of the enzymes that catalyze the β-oxidation of fatty acids, but lower activities of several glycolytic enzymes [48]. Energization of amastigote cells by palmitate plus bicarbonate suggests the β-oxidation of fatty acids, which is relatively more important to Leishmania amastigotes than promastigotes.
The differences between parasite and host energy metabolism described in this paper hold great promise as targets for chemotherapy. For example, most of the active anti-leishmanial compounds tested on promastigotes failed to inhibit amastigotes [49, 50]. If an agent that can specifically inhibit the amastigote fumarate reductase is found, it is expected to be extremely effective and selective as an anti-leishmanial agent.
This work was supported by Indian Council of Medical Research (ICMR), Government of India, Grant 5/8-7(61)2001-ECD-II.
REFERENCES
1.Chang, K. P. (1983) Int. Rev. Cytol. Suppl.,
14, 267-305.
2.Handman, E., Schnur, L. F., Spithill, T. W., and
Mitchell, G. F. (1986) J. Immunol., 137, 3608-3613.
3.Pearson, R. D., and Wilson, M. E. (1989) in
Parasitic Infections in the Compromised Host (Walzer, P. D., and
Genta, R. M., eds.) Marcel Dekker, Inc., New York, pp. 31-81.
4.Ephros, M., Bitnun, A., Shaked, P., Waldman, E.,
and Zilberstein, D. (1999) Antimicrob. Agents Chemother.,
43, 278-282.
5.Ramos, H., Milhaud, J., Cohen, B. E., and Bolard,
J. (1990) Antimicrob. Agents Chemother., 34,
1584-1589.
6.Chen, M., Christensen, S. B., Blom, J., Lemmich,
E., Nadelmann, L., Fich, K., Theander, T. G., and Kharazmi, A. (1993)
Antimicrob. Agents Chemother., 37, 2550-2556.
7.Zilberstein, D., and Shapira, M. (1994) Annu.
Rev. Microbiol., 48, 449-470.
8.Debrabant, A., Joshi, M. B., Pimenta, P. F., and
Dwyer, D. M. (2004) Int. J. Parasitol., 34, 205-217.
9.Barak, E., Amin-Spector, S., Gerliak, E., Goyard,
S., Holland, N., and Zilberstein, D. (2005) Mol. Biochem.
Parasitol., 141, 99-108.
10.Bente, M., Harder, S., Wiesgigl, M., Heukeshoven,
J., Gelhaus, C., Krause, E., Clos, J., and Bruchhaus, I. (2003)
Proteomics, 3, 1811-1829.
11.Brun, R., and Krassner, S. M. (1976) J.
Protozool., 23, 493-497.
12.Rudzinska, M. A., Alesandro, P. A. D., and
Trager, W. (1964) J. Protozool., 11, 166-191.
13.Angelopoulos, E. (1970) J. Protozool.,
17, 39-51.
14.Martin, E., and Mukkada, A. J. (1979) J. Biol.
Chem., 254, 12192-12198.
15.Santhamma, K. R., and Bhaduri, A. (1995) Mol.
Biochem. Parasitol., 75, 43-53.
16.Bera, T., Lakshman, K., Ghanteswari, D., Pal, S.,
Sudhahar, D., Islam, M. N., Bhuyan, N. R., and Das, P. (2005)
Biochim. Biophys. Acta, 1725, 314-326.
17.Biswas, S., Haque, R., Bhuyan, N. R., and Bera,
T. (2008) Biochim. Biophys. Acta, 1780, 116-127.
18.Tielens, A. G., and van Hellemond, J. J. (1998)
Biochim. Biophys. Acta, 1365, 71-78.
19.Hellemond, J. J., van der Klei, A., van Weelden,
S. W., and Tielens, A. G. (2003) Philos. Trans. R. Soc. Lond. B.
Biol. Sci., 358, 205-213.
20.Michels, P. A. M., Michels, J. P. J., Boonstra,
J., and Konings, W. N. (1979) FEMS Microbiol. Lett.,
5, 357-364.
21.Mukhopadhyay, S., Sen, P., Bhattacharyya, S.,
Majumdar, S., and Roy, S. (1999) Vaccine, 17,
291-300.
22.Bera, T. (1987) Mol. Biochem. Parasitol.,
23, 183-192.
23.Berredo-Pinho, M., Peres-Sampaio, C. E.,
Chrispim, P. P., Belmont-Firpo, R., Lemos, A. P., Martiny, A.,
Vannier-Santos, M. A., and Meyer-Fernandes, J. R. (2001) Arch.
Biochem. Biophys., 391, 16-24.
24.Sereno, D., and Lemesre, J. L. (1997)
Antimicrob. Agents Chemother., 41, 972-976.
25.Kar, K., Mukherjee, K., Naskar, K., Bhattacharya,
A., and Ghosh, D. K. (1990) J. Protozool., 37,
277-279.
26.Hauser, R., Pypaert, M., Hausler, T., Horn, E.
K., and Schneider, A. (1996) J. Cell Sci., 109,
517-523.
27.Pedersen, P. L., Greenawalt, J. W., Reynafarje,
B., Hullihen, J., Decker, G. L., Soper, J. W., and Bustamente, E.
(1978) Meth. Cell Biol., 20, 411-481.
28.Robinson, J., and Cooper, J. M. (1970) Anal.
Biochem., 33, 390-399.
29.Avron, M., and Shavit, N. (1963) Anal.
Biochem., 6, 549-554.
30.Pecci, L., Montefoschi, G., Fontana, M., and
Cavallini, D. (1994) Biochem. Biophys. Res. Commun., 199,
755-760.
31.Singer, T. P. (1974) Meth. Biochem.
Anal., 22, 123-175.
32.Rieske, J. S. (1967) in Methods in
Enzymology (Colowick, S. P., and Kaplan, N. O., eds.) Vol. X,
Academic Press, New York-London, p. 240.
33.Bril, C. (1954) Biochim. Biophys. Acta,
15, 258-262.
34.Vercesi, A. E., and Docampo, R. (1992)
Biochem. J., 284, 463-467.
35.Akerman, E. O., and Jarvisalo, J. O. (1980)
Biochem. J., 192, 183-190.
36.Gornall, A. G., Bardawill, C. J., and Daird, M.
M. (1949) J. Biol. Chem., 177, 751-766.
37.Markwell, M. K., Hass, S. M., Bieber, L. L., and
Tolbert, N. E. (1978) Anal. Biochem., 87, 206-210.
38.Datta, G., and Bera, T. (2001) Biochim.
Biophys. Acta, 1512, 149-157.
39.Datta, G., and Bera, T. (2002) J. Eukaryot.
Microbiol., 49, 24-29.
40.James, P. E., Grinberg, O. Y., and Swartz, H. M.
(1998) J. Leukoc. Biol., 64, 78-84.
41.McConville, M. J., de Souza, D., Saunders, E.,
Likic, V. A., and Naderer, T. (2007) Trends Parasitol.,
23, 368-375.
42.Huttunen, M. T., and Akerman, K. E. (1980)
Biochim. Biophys. Acta, 597, 274-284.
43.Vercesi, A. E., Bernardes, C. F., Hoffmann, M.
E., Gadelha, F. R., and Docampo, R. (1991) J. Biol. Chem.,
266, 14431-14434.
44.Akerman, K. E. (1979) Biochim. Biophys.
Acta, 546, 341-347.
45.Martin, E., Simon, M. W., Schaefer, F. W., 3rd.,
and Mukkada, A. J. (1976) J. Protozool., 23, 600-607.
46.Rainey, P. M., and MacKenzie, N. E. (1991)
Mol. Biochem. Parasitol., 45, 307-315.
47.Assaily, W., and Benchimol, S. (2006) Cancer
Cell, 10, 4-6.
48.Coombs, G. H., Craft, J. A., and Hart, D. T.
(1982) Mol. Biochem. Parasitol., 5, 199-211.
49.Mattock, N. M., and Peters, W. (1975) Ann.
Trop. Med. Parasitol., 69, 359-371.
50.Peters, W., Trotter, E. R., and Robinson, B. L.
(1980) Ann. Trop. Med. Parasitol., 74, 321-335.