A Powerful Hybrid puc Operon Promoter Tightly Regulated by Both IPTG and Low Oxygen Level
Zongli Hu#, Zhiping Zhao#, Yu Pan, Yun Tu, and Guoping Chen*
Key Laboratory of Biorheological Science and Technology and Bioengineering College, Chongqing University, 174 Shapingba Main Street, Chongqing 400044, People’s Republic of China; fax: (0086) 23-6511-2674; E-mail: chenguoping@cqu.edu.cn; huzongli@cqu.edu.cn; zhipingzhao@cqu.edu.cn; ipying@163.com; tuyun@cqu.edu.cn# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received September 8, 2009; Revision received November 5, 2009
Rhodobacter sphaeroides has been intensively studied and provides an excellent model for studying both photosynthesis and membrane development. The photosynthetic apparatus (LH2 and LH1–RC complexes) can be synthesized in large scale and integrated into the intracytoplasmic membrane system under specific conditions, which thus provides us insight to utilize the puc or(and) puf operon to heterologously express recombinant proteins in the intracytoplasmic membrane using Rb. sphaeroides as a novel expression system. However, basal level of expression of puc and puf promoter is uncontrolled. We report the construction of LH2 polypeptide expression vector that contains a reengineered lacIq-puc promoter–lac operator hybrid promoter, which allows the puc operon to be regulated by both IPTG and low oxygen level. Synthesis of LH2 complexes was completely repressed in the absence of isopropyl β-D-thiogalactoside (IPTG), and the degree of induction was controlled by varying the concentration of IPTG. The optimal concentration of IPTG was determined. SDS-PAGE and Western blot were employed for further analysis. Our results suggest that the reengineered hybrid promoter is efficient to tightly regulate the expression of the puc operon, and our strategy can open up a new approach in the study of the membrane protein expression system.
KEY WORDS: LH2, hybrid promoter, IPTG, Rhodobacter sphaeroidesDOI: 10.1134/S0006297910040176
Abbreviations: ICM, intracytoplasmic membrane; IPTG, isopropyl β-D-thiogalactoside; LH1(2), light-harvesting complex 1(2); RC, reaction center.
Rhodobacter sphaeroides is a purple non-sulfur gram-negative
facultative anaerobe capable of anoxygenic photosynthesis [1]. It has multiple growth models including aerobic
and anaerobic respiration, fermentation, and anoxygenic photosynthesis
[2]. It is also one of the most diverse subgroups
of the α-3 subgroup of the Proteobacteria [3]. This bacterium can grow under both
photoheterotrophic and chemoheterotrophic conditions. When growing
chemoheterotrophically, Rb. sphaeroides has a typical
gram-negative cell envelope structure and its growth is supported by
aerobic respiration. When oxygen is removed from such a culture, a
series of events is triggered which, through a process of invagination,
results in the differentiation of the cytoplasmic membrane into
specialized domains and the formation of the intracytoplasmic membrane
(ICM) system, which houses the photosynthetic apparatus. The
proliferation of this membrane provides an increased surface area for
the absorption and utilization of light by the photosynthetic
apparatus. The photosynthetic apparatus consists of three
membrane-bound pigment–protein complexes. Light-harvesting
complexes 1 and 2 (LH1 and LH2), which are the major
pigment–protein complexes, have been designated B800-850 and B875
based on their near-infrared absorption maxima [4].
The third complex incorporates reaction center (RC) along with the
associated components required for subsequent electron transport and
energy transduction. The ratio of LH1 to RC is fixed at approximately
15 : 1, whereas the ratio of LH2 to the LH1–RC unit is variable,
changing in a manner inverse to light intensity [5].
It has been well reported that LH2 and LH1 complexes are comprised of α- and β-polypeptide with highly symmetric arrangement, binding noncovalently bacteriochlorophylls and carotenoids, and are constructed in a remarkably similar fashion. However, RC is comprised of three subunits with unsymmetrical arrangement, namely H, L, and M [6]. The LH2 complex from Rb. sphaeroides is a nonamer [7], like many other purple bacteria, such as Rhodopseudomonas acidophila and Rhodovulum sulfidophilum [8]. However, LH2 from Rhodospirillum molischianum is an octamer [9, 10]. In the nonamers, each unit contains one α/β polypeptide, the nine α-polypeptides form a hollow cylinder with the nine β-polypeptides arranged outside. LH2 β-polypeptide and α-polypeptide of Rb. sphaeroides are encoded by the pucB and pucA, respectively. It has been well-documented that photosynthesis genes including bch, crt, pucBA, pufBA, pufLM, puhA, etc. are tightly regulated by oxygen tension and light intensity with uncertain mechanisms. Oxygen can shut down the synthesis of photosynthetic apparatus very rapidly. Similarly, photosynthesis genes are also regulated inversely to the incident light intensity [11].
Rhodobacter sphaeroides is the best-characterized purple bacterium and provides an ideal model system for studying both photosynthesis and membrane development [2]. Additionally, LH2 and LH1–RC complexes account for a major portion of the ICM, and they can be synthesized in large scale and then integrated into the ICM system under anaerobic conditions. These provide us insight to utilize the puc or(and) puf operon to heterologously express recombinant proteins in ICM using Rb. sphaeroides as a novel expression system. As we have described above, photosynthesis genes are regulated by oxygen tension. Unfortunately, in many cases the LH2 and LH1–RC complexes can still be synthesized at basal level even in the presence of oxygen. This property can be particularly troublesome when utilize the puc or(and) puf operon to express recombinant proteins or other gene products that are toxic to the cell using Rb. sphaeroides as a platform. Experiments requiring strong repression and precise control of cloned genes can be difficult to conduct because of the relatively high basal level of expression of currently employed promoters [12], such as puc and puf promoter of Rb. sphaeroides [13]. However, vast numbers of strong hybrid promoters as well as some of their derivatives have been constructed and widely applied for expression of genes of interest typically based on the E. coli lac operon [14], which provides us clear insight to construct a hybrid promoter to regulate the expression of a gene of interest in Rb. sphaeroides. LacI repressor is one of the best-studied prokaryotic transcriptional regulatory proteins, which can bind to lac operator (lacO) and prevent the RNA polymerase from transcribing three structural genes – lacZ, lacY, and lacA. The puc operon of Rb. sphaeroides consists of the puc promoter, pucBA genes encoding LH2 β- and α-polypeptides, respectively, and downstream pucC gene helping to assemble the LH2 complexes into ICM. Here we construct an LH2 polypeptide expression vector with a powerful hybrid lacIq-puc promoter-lac operator allowing dual regulation by both isopropyl β-D-thiogalactoside (IPTG) and low oxygen level.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions. Strains and plasmids used in this study are listed in the table. Rhodobacter sphaeroides strains were grown at 34°C in M22+ medium supplemented with 0.1% casamino acids for growth in liquid culture [20]. Growth under low oxygen tension was performed by incubating 750 ml of culture in 1-liter flasks under gentle agitation. For aerobic cultivation, 200 ml cultures were vigorously shaken in 1-liter flasks.
Escherichia coli strains were grown aerobically at 37°C in Luria–Bertani (LB) medium. Antibiotics were added to the growth media at the following concentrations: ampicillin, 100 µg/ml for E. coli; tetracycline, 10 µg/ml for E. coli and 1 µg/ml for Rb. sphaeroides; neomycin, 20 µg/ml for Rb. sphaeroides; streptomycin, 5 µg/ml for Rb. sphaeroides.
Constructions of plasmid vectors. PCRs were carried out using PrimeSTARTM HS DNA polymerase from TAKARA. Primers used in this study are listed in the table. All amplified fragments were ligated into the pUC19 cloning vector and sequenced. For the construction of pRKlacIqpucPpucBHis10AC expression vector, PCR product containing lacIq amplified from E. coli JM109 was cut from pUC19-lacIq with EcoRI and SacI and ligated into the pRK415 vector cut with the same restriction enzymes, producing plasmid pRK-lacI. Then the PCR product containing the pucP promoter and lacO was removed from pUC19-pucP-lacO vector digested with XhoI and KpnI and ligated into pRK-lacI between the XhoI and KpnI sites, producing plasmid pRK-PO. After that, PCR product containing SD sequence, pucB, and Xa factor protease recognition site was cut from pUC19-SD-pucB-Xa with KpnI and XbaI and subsequently ligated into the pRK-PO vector digested with the same restriction enzymes, producing plasmid pRK-BXa. Finally, PCR product containing His10-tag, pucA, pucC, and terminator was removed from pUC19-His10-pucA-pucC-terminator vector and ligated into the pRK-BXa vector, both vectors digested with XbaI and PstI, producing plasmid pRKlacIqpucPpucBHis10AC. Plasmid pRKpucPpucBHis10AC was constructed in a similar way.
Bacterial strains and plasmids used in this study
Conjugation techniques. Plasmid DNA was mobilized into Rb. sphaeroides by using E. coli S17-1 as the donor as described previously [20]. Transconjugants were grown aerobically in the dark on plates of M22+ medium supplemented with appropriate antibiotics as described above.
Spectroscopy. Spectra of whole cells were recorded on a Perkin Elmer lambda 900 UV/VIS spectrometer (USA). A suitable blank of growth medium was used in each case.
Expression of LH2 complexes. Cell cultures were shifted from aerobic conditions to semi-aerobic conditions at A600 of 0.5-0.8, and continued incubation for about 8 h after adding IPTG.
Protein purification and Western blot analysis. Cells were resuspended in 20 mM Tris buffer (pH 8.0) in the presence of 100 mM NaCl and 1 mM phenylmethylsulfonyl fluoride and subsequently broken by three passages through a high-pressure homogenizer (Italy) at 1200 bar. Membranes were resuspended with 1.0% (v/v) lauryldimethylamine N-oxide and purified by Ni-IDA. For Western blot analysis, proteins were separated on 15-20% SDS-PAGE gradient gel and transferred to PVDF membrane. Anti-His antibody and goat anti-rabbit IgG-alkaline phosphatase were used as primary and secondary antibody, respectively.
RESULTS
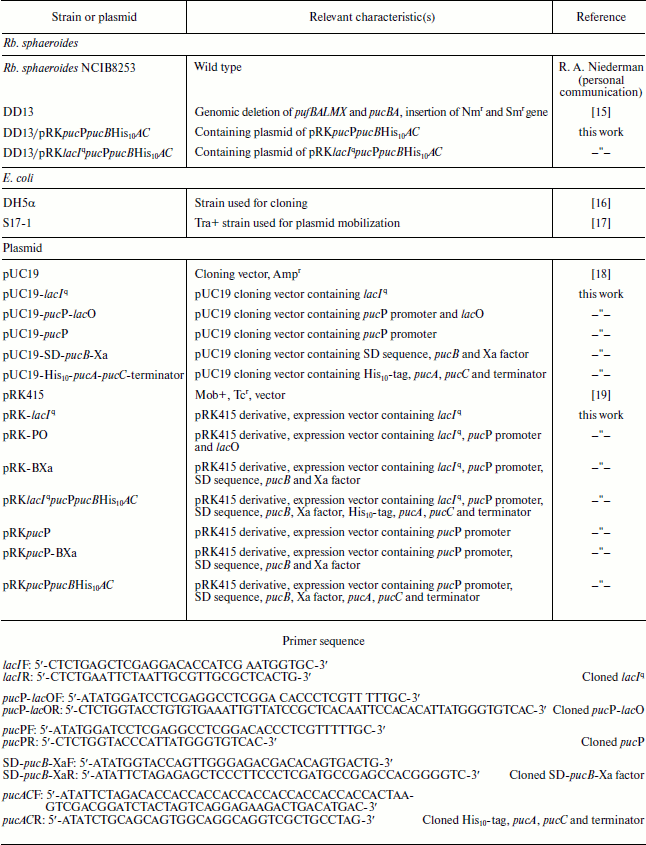
Constructions of expression vectors. The resulting expression vectors were termed pRKlacIqpucPpucBHis10AC and pRKpucPpucBHis10AC, as shown in Fig. 1. Both expression vectors are based on pRK415, which is a broad-range host plasmid. Figure 1a represents the construction of pRKlacIqpucPpucBHis10AC expression vector containing the powerful hybrid promoter. Construction of pRKpucPpucBHis10AC carrying native promoter pucP only is depicted in Fig. 1b. Genes encoding LH2 complexes in pRKlacIqpucPpucBHis10AC can be expressed under the tight control of the hybrid promoter. The β- and α-polypeptide encoded by pucB and pucA will be integrated into the ICM by pucC encoding protein in both DD13/pRKlacIqpucPpucBHis10AC and DD13/pRKpucPpucBHis10AC strains. Because of the introduction of the lacIq and lacO into pucP promoter, respective inducer should be taken into account to derepress the LacIq repressor protein when the puc operon is expressed. Apparently, genes of interest could be inserted into both vectors at the SacI and XbaI sites to construct recombinant proteins, which will be expressed, then integrated into the ICM following the native membrane protein under the control of pucC encoding protein. There is a Xa factor protease recognition sequence and a His10-tag upstream and downstream of the SacI and XbaI sites, respectively.
Expression of LH2 complexes and determination of optimal IPTG concentration. With conjugation techniques, plasmid pRKlacIqpucPpucBHis10AC was transferred into Rb. sphaeroides mutant DD13. When cell cultures of DD13 transconjugants reach OD600 of 0.5-0.8, IPTG is added to the growth medium and simultaneously the cell cultures are shifted from aerobic conditions to semi-aerobic conditions. After that, cell cultures of DD13 transconjugants are scanned at the band of 700-900 nm. Two spectral peaks appear at ~800 and ~850 nm, suggesting the production of LH2 complexes, and the spectra are scaled to reflect the level of LH2 complexes per amount of cell cultures, as shown in Fig. 2. The degree of induction is controlled by varying the concentration of IPTG. Growth without IPTG allowed no background expression (Fig. 2d), while growth with concentrations of IPTG at 0.1 and 0.5 mM resulted in low-level production of LH2 complexes (Fig. 2, a and b). In contrast, IPTG at the concentrations of 0.8, 1.0, and 1.5 mM was sufficient to activate the hybrid promoter and consequently yield high level production of LH2 complexes (Fig. 2, c, e, and f), and 1.0 mM is considered as the optimal concentration in subsequent studies.Fig. 1. Schematic representation of the expression vector constructs. a) The plasmid pRKlacIqpucPpucBHis10AC contains the hybrid promoter comprised of E. coli lacIq and lacO, pucP promoter of Rb. sphaeroides and structural genes pucB and pucA encoding β- and α-polypeptide of LH2 complex. b) Plasmid pRKpucPpucBHis10AC lacking the E. coli lacIq and lacO covers the same structure compared to that of the pRKlacIqpucPpucBHis10AC. Start and Stop represents start codon and stop codon, respectively.
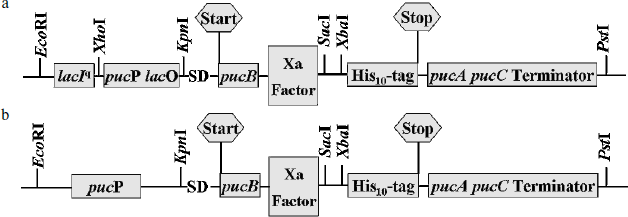
Because the hybrid promoter we constructed allows a dual regulation by both IPTG and low oxygen level, only meeting the two requirements can result in the production of LH2 complexes. Clearly, there is no background synthesis of LH2 complexes under uninduced conditions (without IPTG) even for cells grown under semi-aerobic conditions, as shown in Figs. 2d and 3a. Similarly, no LH2 complexes were produced when cells grown under aerobic conditions even in the presence of IPTG, as shown in Fig. 3b. Cell cultures of DD13/pRKpucPpucBHis10AC showed similar spectral properties compared to that of DD13/pRKlacIqpucPpucBHis10AC, as revealed in Fig. 3c.Fig. 2. Spectral properties of DD13/pRKlacIqpucPpucBHis10AC cell cultures after adding IPTG to the growth medium. Low concentrations of IPTG (0.1 mM (a), 0.5 mM (b)) cannot completely activate the hybrid promoter, while high concentrations of IPTG (0.8 mM (c), 1.0 mM (e), 1.5 mM (f)) probably activate the hybrid promoter completely. Growth without IPTG (0 mM (d)) allowed no background expression.
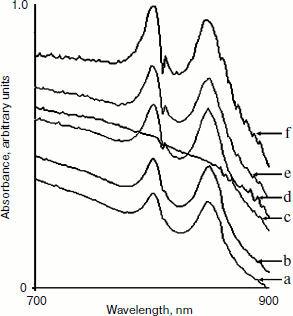
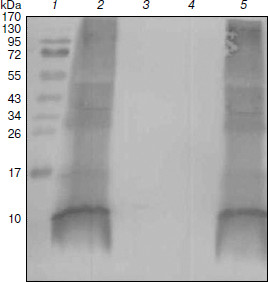
Purification and identification of the expressed protein. To further test our strategy, we purified the expressed protein from Rb. sphaeroides DD13 transconjugants by Ni-IDA. The resulting purified protein was examined by SDS-PAGE, and a band of approximately 10 kDa was observed on Coomassie Brilliant Blue-stained 15-20% SDS-polyacrylamide gradient gel, as revealed in Fig. 4. Western blot analysis was performed using anti-His antibody. As shown in Fig. 5, a band of approximately 10 kDa was observed on PVDF membrane. All this reliable evidence suggested the hybrid promoter we constructed was efficient to tightly regulate the expression of target genes, and our strategy actually worked well.Fig. 3. Spectral absorbance of DD13/pRKlacIqpucPpucBHis10AC (a, b) and DD13/pRKpucPpucBHis10AC (c, d). a, b) Absorbance of DD13/pRKlacIqpucPpucBHis10AC grown under semi-aerobic conditions in the absence of IPTG (a) and under aerobic conditions in the presence of IPTG (b). c, d) Absorbance of DD13/pRKpucPpucBHis10AC grown under aerobic conditions in the absence of IPTG (c) and under semi-aerobic conditions in the absence of IPTG (d).
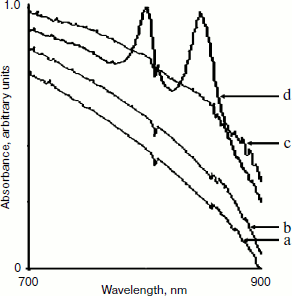
Fig. 4. SDS-PAGE analysis of purified proteins. Lanes: 1) molecular weight markers; 2) protein purified from DD13/pRKlacIqpucPpucBHis10AC with IPTG induction; 3) protein purified from DD13/pRKpucPpucBHis10AC; 4) sample purified from DD13 as control; 5) sample purified from DD13/pRKlacIqpucPpucBHis10AC without IPTG induction.
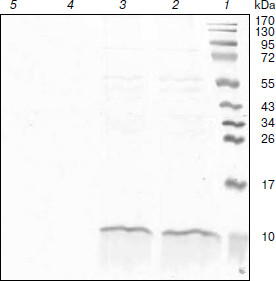
Fig. 5. Western blot analysis of purified proteins. Lanes: 1) molecular weight markers; 2) protein purified from DD13/pRKlacIqpucPpucBHis10AC with IPTG induction; 3) sample purified from DD13/pRKlacIqpucPpucBHis10AC without IPTG induction; 4) sample purified from DD13 as control; 5) protein purified from DD13/pRKpucPpucBHis10AC.
DISCUSSION
It is well known that the lac operon plays important roles in the development of molecular and systems biology [21], and it displays much of the complexity and subtlety inherent in gene regulation, including transcription, translation, protein assembly, protein degradation, binding of different proteins to DNA, and binding of small molecules to DNA-binding proteins [22]. The lac operon consists of a regulatory domain and three structural genes with related functions required for the uptake and catabolism of lactose. A regulatory protein, the LacI repressor, is a homotetramer comprised of two functional homodimers [23, 24], and it is one of the best-studied prokaryotic transcriptional regulatory proteins [25]. It can bind to lacO and prevent the RNA polymerase from transcribing the three structural genes. The gene lacIq is a mutant lacI that produces 10-fold more repressor than lacI is often used in recombinant strains to provide better repression of recombinant gene expression in the absence of inducer [26]. Induction of the lac operon occurs only when the inducer molecule binds to the repressor, and the inducers that can be used include allo-lactose, a metabolic product of lactose and gratuitous inducers, such as IPTG or methyl-β-D-thiogalactoside, and we selected IPTG to derepress the negative regulation of LacIq repressor in this study. The probability for the inducer to bind to the repressor depends on the inducer concentration inside the cell. The puc operon of Rb. sphaeroides is composed of the pucP promoter and pucBA genes encoding the β- and α-polypeptides of the LH2 complex, respectively. Additional downstream open reading frame(s) are present that encode gene product(s) involved in the posttranscriptional control of expression and assembly of the β- and α-polypeptides to form the LH2 complexes [27].
In this study, we have successfully constructed LH2 polypeptide expression vector carrying a hybrid promoter comprised of lacIq and lacO of E. coli JM109 and puc operon promoter pucP of Rb. sphaeroides. Spectral properties, SDS-PAGE, and Western blot analysis indicate that the hybrid promoter actually works well. The puc operon promoter pucP can be greatly repressed by high oxygen level. Despite the negative regulation by high oxygen tension, IPTG plays a crucial role in the production of LH2 complexes. Thus, we can get the spectral properties of cultures of Rb. sphaeroides DD13 transconjugants grown under semi-aerobic conditions after adding IPTG. As expected, there is no background expression of LH2 complexes without IPTG even under semi-aerobic conditions, as shown in Fig. 3a. Similarly, we cannot obtain spectral properties of cultures grown under aerobic conditions even after adding IPTG (Fig. 3b).
In addition, there is no background expression without IPTG in Rb. sphaeroides DD13 transconjugants. This property can be used to overexpress proteins that are lethal to the cells. On the other hand, in cell cultures of transconjugants, two absorption bands can be observed at ~800 and ~850 nm after induction, which can be used as the marker for harvest of the cells. With the His10-tag in the C-terminus of pucB, the recombinant proteins can be purified by the affinity tag and identified by Western blot. Actually, the His10-tag can also be fused at the C-terminus of pucA, which will achieve the same goal. It is worth being determined that if there is an expected band in the Western blot, the Xa factor can be used to cut off pucB encoding protein to obtain more highly purified target proteins. The next step is to utilize the LH2 expression vector bearing the hybrid promoter to express recombinant proteins using Rb. sphaeroides as a novel host.
We thank Prof. C. Neil Hunter for the DD13 strain.
This work was supported by the program “863” of the Ministry of Science and Technology of the People’s Republic of China (No:2006AA02Z138) and National Natural Science Foundation of China (No:30771463, 30871709).
REFERENCES
1.Naylor, G. W., Addlesee, H. A., Gibson, L. C. D.,
and Hunter, C. N. (1999) Photosynth. Res., 62,
121-139.
2.Kiley, P. J., and Kaplan, S. (1988) Microbiol.
Rev., 52, 50-69.
3.Woese, C. R., Stackebrandt, E., Weisburg, W. G.,
Paster, B. J., Madigan, M. T., Fowler, V. J., Hahn, C. M., Blanz, P.,
Gupta, R., Nealson, K. H., and Fox, G. E. (1984) Syst. Appl.
Microbiol., 5, 315-326.
4.F. J.a, E., E..Grondelle, R. V.Boonstra, A. F.,
Visschers, R. W., Calkoen, F., van Grondelle, R., van Bruggen, E. F.
J., and Boekema, E. J. (1993) Biochim. Biophys. Acta,
1142, 181-188.
5.Zeilstra-Ryalls, J., Gomelsky, M., Eraso, J. M.,
Yeliseev, A., O’Gara, J., and Kaplan, S. (1998) J.
Bacteriol., 180, 2801-2809.
6.Hu, X., Ritz, T., Damjanovic, A., and Autenrieth,
F. (2002) Q. Rev. Biophys., 35, 1-62.
7.Walz, T., Jamieson, S. J., Bowers, C. M., Bullough,
P. A., and Hunter, C. N. (1998) J. Mol. Biol., 282,
833-845.
8.Savage, H., Cyrklaff, M., Montoya, G., Kuhlbrandt,
W., and Sinning, I. (1996) Structure, 4, 243-252.
9.Klinekofort, W., Germeroth, L., van den Borek, J.
A., Schubert, D., and Michel, H. (1992) Biochim. Biophys. Acta,
1140, 102-104.
10.Koepke, J., Hu, X., Muenke, C., Schulten, K., and
Michel, H. (1996) Structure, 4, 581-597.
11.Pemberton, J. M., Horne, I. M., and McEwan, A. G.
(1998) Microbiology, 144, 267-278.
12.Khan, S. R., Gaines, J., Roop, R. M., 2nd., and
Farrand, S. K. (2008) Appl. Environ. Microbiol.,
74, 5053-5062.
13.Oh, J. I., and Kaplan, S. (2000) EMBO J.,
19, 4237-4247.
14.Mardanov, A. V., Strakhova, T. S., Smagin, V. A.,
and Ravin, N. V. (2007) Gene, 395, 15-21.
15.Jones, M. R., Fowler, G. J., Gibson, L. C.,
Grief, G. G., Olsen, J. D., Crielaard, W., and Hunter, C. N. (1992)
Mol. Microbiol., 6, 1173-1184.
16.Eraso, J. M., and Kaplan, S. (1994) J.
Bacteriol., 176, 32-43.
17.Simon, R., Priefer, U., and Puhler, A. (1983)
Biotechnology (NY), 1, 784-791.
18.Yanisch-Perron, C., Vieira, J., and Messing, J.
(1985) Gene, 33, 103-119.
19.Keen, N. T., Tamaki, S., Kobavashi, D., and
Trollinger, D. (1988) Gene, 70, 191-197.
20.Hunter, C. N., and Turner, G. (1988) J. Gen.
Microbiol., 134, 1471-1480.
21.Santillan, M., and Mackey, M. C. (2008) J. R.
Soc. Interface, 5, S29-S39.
22.Vilar, J. M., Guet, C. C., and Leibler, S. (2008)
J. Cell Biol., 161, 471-476.
23.Lewis, M. (2005) C. R. Biol., 328,
521-548.
24.Wilson, C. J., Zhan, H., Swint-Kruse, L., and
Matthews, K. S. (2008) Cell. Mol. Life Sci., 64,
3-16.
25.Lakshmi, O. S., and Rao, N. M. (2009) Protein
Eng. Des. Sel., 22, 53-58.
26.Donovan, R. S., Robinson, C. W., and Glick, B. R.
(1996) J. Ind. Microbiol., 16, 145-154.
27.Lee, J. K., Kiley, P. J., and Kaplan, S. (1989)
J. Bacteriol., 171, 3391-3405.