Promyelocytic Leukemia Protein Interacts with Werner Syndrome Helicase and Regulates Double-Strand Break Repair in γ-Irradiation-Induced DNA Damage Responses
Jilai Liu#, Yi Song#*, Junjie Qian, Bin Liu, Yan Dong, Baolei Tian, and Zhixian Sun*
Department of Biochemistry and Molecular Biology, Beijing Institute of Radiation Medicine, 27 Taiping Road, Beijing 100850, P. R. China; fax: 86-10-6817-7417; E-mail: songyibj@sina.com.cn; sunzx@nic.bmi.ac.cn# These authors contributed equally to this work.
* To whom correspondence should be addressed.
Received October 18, 2010; Revision received December 30, 2010
We show here that γ-irradiation leads to the translocation of endogenous Werner syndrome helicase (WRN) from nucleoli to nucleoplasmic DNA double strand breaks (DSBs), and WRN plays a role in damage repair. The relocation of WRN after irradiation was perturbed by promyelocytic leukemia protein (PML) knockdown and enhanced by PML IV overexpression. PML IV physically interacted with WRN after irradiation. Amino acids (a.a.) 394 to 433 of PML were necessary for this interaction and the nucleoplasmic translocation of WRN and were involved in DSB repair and cellular sensitivity to γ-irradiation. Taken together, our results provide molecular support for a model in which PML IV physically interacts with and regulates the translocation of WRN for DNA damage repair through its 394-433 a.a. domain.
KEY WORDS: PML, WRN, γ-irradiation, DNA damage, DNA repairDOI: 10.1134/S000629791105004X
Abbreviations: ATM, ataxia-telangiectasia mutated protein; DAPI, 4′,6-diamidino-2-phenylindol; DSB, double-strand break; GFP, green fluorescent protein; γ-H2AX, phosphorylated histone H2AX; HEL, human embryo lung fibroblasts; IRIF, ionizing radiation induced foci; PML, promyelocytic leukemia protein; PML NBs, promyelocytic leukemia nuclear bodies; WRN, Werner syndrome helicase; WS, Werner’s syndrome.
Werner syndrome helicase (WRN), the protein deficient in the autosomal
recessive disorder Werner syndrome (WS), is hypothesized to function in
various DNA metabolic pathways by analogy with RecQ DNA helicase. One
of the major hallmarks of WS is genomic instability, indicating that
WRN is required for the maintenance of genomic integrity [1]. It has been reported that WRN is translocated from
nucleoli to the nucleoplasm in DNA damage responses and is involved in
the repair of DNA damage in mammalian cells [2-4].
Promyelocytic leukemia nuclear bodies (PML NBs) are dynamic macromolecular structures that respond to diverse DNA damage stresses by altering their number, size, and content. The dynamic behavior of PML NBs is related to the processes of fundamental cellular defense including DNA repair, transcription, and apoptosis. It has been reported that many repair factors are translocated to or from PML NBs during DNA damage responses [5]. Though it has been reported that ectopically expressed WRN can form nucleoplasmic foci and partially colocalize with PML NBs, the functional relationship between endogenous PML (promyelocytic leukemia protein) and WRN remains undefined [6, 7].
Here, we demonstrate that WRN translocates from the nucleoli to the nucleoplasm and colocalizes with the irradiation-induced γ-H2AX and phosphorylated ATM foci following γ-irradiation, and is critical for DSB repair. Endogenous PML is physically associated with WRN, and amino acids 394-433 of PML are required for this interaction and double-strand break (DSB) repair. These data suggest that PML plays a role in signaling or in facilitating DNA repair in cooperation with WRN.
MATERIALS AND METHODS
Construction of plasmids. The PML IV expression vectors pEGFP-PML
and pCMV-myc-PML carry the PML transcript corresponding to human PML
IV, as previously described [8]. The truncation and
deletion plasmids of PML IV were constructed by PCR with primers listed
below (several bases at the 5′ end, added for endoenzyme
digestion, are not shown):
PML IV F1 (1-633 a.a.):
5′-ATGGAGCCTGCACCCGCCCGATCTCCGAG-3′;
PML IV F2 (361-633 a.a):
5′-CAGGAGGAGCCCCAGAGCCT-3′;
PML IV F3 (394-633 a.a.):
5′-GATGCAGCTGTATCCAAGAA-3′;
PML IV F4 (433-633 a.a.):
5′-GCTGAAGCCCAGCCTATGGCT-3′;
PML IV F4 (delete 433-633 a.a.):
5′-AGCTCTTGCATCACCCAGGGGGCTGAAGCCCAGCCTATGGCT-3′;
PML IV R1 (1-360 a.a.):
5′-TTAGCGCAGGCGGCAGAGCGCCT-3′;
PML IV R2 (delete 433-633 a.a.):
5′-AGCCATAGGCTGGGCTTCAGCCCCCTGGGTGATGCAAGAGCT-3′;
PML IV R3 (1-633 a.a.):
5′-CTAAATTAGAAAGGGGTGGGGGTA-3′.
Cell culture and gene transfection. The primary human embryo lung fibroblasts (HEL) (a generous gift from Dr. Xiao-fei Zheng), breast carcinoma cell line (MCF-7), and lung adenocarcinoma epithelial cells (A549) were cultured in Dulbecco’s modified Eagle’s medium (Gibco, USA) containing 10% fetal bovine serum. Plasmid transfection was conducted with the FuGENE HD Transfection Reagent (Roche Diagnostics, Germany).
The target sequence of siRNA for PML (5′-GAGUCGGCCGACUUCUGGU-3′) and WRN (5′-GUUCUUGUCACGUCCUCUG-3′) are from previous studies [9, 10]. These were synthesized by GenePharma Company (China). Proliferating cells were transfected with the siRNA by INTERFERin siRNA transfection reagent (Polyplus-transfection, France).
Immunoprecipitation. Cells (1·107) were lysed in M-PER buffer (Invitrogen, USA) with protease inhibitor cocktails (Roche, Germany). Then, 500 µg total protein and 5 µg antibody were used in the coprecipitation experiments. Pre-cleared cell lysates were incubated with antibody and gently agitated for at least 1 h at 4°C. Next, 20 μl protein A/G Plus-agarose beads (Santa Cruz, USA) were added and incubated overnight at 4°C. The beads were collected and washed extensively with lysis buffer five times. Immune complexes were released from the beads by boiling in 2× SDS-PAGE sample buffer. Tag antibodies (anti-myc and anti-GFP) used in the precipitation were purchased from MBL company (Japan).
Immunofluorescence staining. Cells were grown on glass coverslips in 12-well plates. At the indicated time after radiation (10 Gy, using 60Co source emitting at a dose rate of 3.66 Gy/min), samples were washed with phosphate-buffered saline three times and fixed with ice-cold 4% paraformaldehyde for 20 min, followed by a 10 min incubation with 0.2% Triton X-100. The coverslips were blocked for 60 min with 1% bovine serum albumin in PBS. Cells were incubated with primary antibody (WRN H-300, PML H238, or C23 from Santa Cruz; pATM and γ-H2AX from Cell Signaling (USA)) for 15 h at 4°C, washed three times with PBS, and incubated with secondary antibody (Alexa 555- or Alexa 488-labeled) for 1 h at room temperature. All fluorescent images were obtained using a Carl Zeiss 510 (Germany) confocal laser-scanning microscope (CLSM) or an Olympus IX-70 (Japan) microscope. Images of the same antibody staining were obtained using the same CLSM parameters (brightness, contrast, pinhole, etc.). Three to six independent experiments were conducted.
Western blot. For western blot analysis, cells were lysed in ice-cold RIPA (radioimmune precipitation assay) buffer containing a complete protease inhibitor cocktail (Roche). After 1 h of gentle stirring at 4°C, samples were centrifuged at 12,000g at 4°C for 20 min to remove insoluble material. After normalizing for protein concentration (BCA Protein Assay Kit; Pierce, USA), 40 µg of lysate was separated by 10% SDS-polyacrylamide gels, transferred to Hybond-ECL (GE Healthcare, USA) and visualized by incubation with various primary antibodies and peroxidase-conjugated secondary antibodies (Invitrogen, USA). WRN (H-300; Santa Cruz) and PML (E15; Santa Cruz) antibodies were used for immunoblotting analysis.
Apoptosis analysis. Cells were washed with PBS. FITC-Annexin V was diluted to a concentration of 1 mg/ml in binding buffer. Cells were resuspended in 1 ml of this solution and incubated for 10 min in the dark at room temperature. Propidium iodide was added into to the cell suspension prior to analysis to give a final concentration of 1 mg/ml. The cells were then examined by flow cytometry. For morphological evaluation of apoptosis, cells were fixed with 4% paraformaldehyde, stained with 1 μg/ml DAPI (′,6-diamidino-2-phenylindol) dye for 5 min, and examined under the fluorescence microscope (Olympus, Japan).
RESULTS
WRN translocates to the nucleoplasm and functions in DSB repair after γ-irradiation. We first investigated the dynamics of WRN expression and localization during the γ-irradiation-induced DNA damage response in HEL cells by immunofluorescence and Western blot. WRN was mainly localized in the nucleoli of untreated cells and was translocated into the nucleoplasm after irradiation (Fig. 1a (see color insert) and Fig. S1c (see Supplement to the article on site of Biochemistry (Moscow) http://protein.bio.msu.ru/biokhimiya), similar to previous reports in other cell types [2, 4]. To assess the nucleoplasmic translocation of WRN semi-quantitatively, HEL cells with nucleoplasmic WRN were scored before and after irradiation. About 10% of unirradiated cells had nucleoplasmic WRN, and the proportion of cells with nucleoplasmic WRN foci significantly increased and peaked 6 h after irradiation. As shown in Fig. 1b, more than 65% of the cells displayed nucleoplasmic WRN localization 6 h after irradiation. The overall protein level of WRN increased slightly during this period (Fig. 1c). Additionally, we analyzed the cell cycle of irradiation-treated HEL cells by flow cytometry. S phase cells were not increased until 9 h after irradiation, suggesting that the nucleoplasmic relocalization of WRN was a damage-associated event and was not due to changes in the cell cycle (Fig. S2 in Supplement).
To determine whether WRN localizes at the irradiation-induced DSB sites, double color immunofluorescence with antibodies against WRN and two important DSB response proteins (γ-H2AX and ATMser1981-p) was performed. At 6 h after irradiation, partial nucleoplasmic WRN foci were observed at sites of γ-H2AX and ATMser1981-p foci (Fig. 1d), indicating that WRN was associated with the irradiation-induced DNA DSB repair foci. The rate of disappearance of radiation-induced γ-H2AX correlates with the rate of DNA repair [11]. We analyzed the kinetics of γ-H2AX clearance after irradiation in WRN knockdown cells and control cells to evaluate the function of WRN in DSB repair. In control cells, the γ-H2AX foci were reduced 3 h after irradiation; however, WRN-knockdown cells displayed more γ-H2AX foci (Fig. 1e), suggesting that DSB repair was perturbed in WRN knockdown cells and translocation of WRN may be necessary for DSB repair.Fig. 1. WRN translocated to the nucleoplasm following irradiation and is involved in irradiation-induced DSB repair. a) Dynamics of WRN expression after γ-irradiation. HEL cells were fixed 6 h after irradiation, and antibodies against WRN and C23 (nucleoli marker) were used for immunofluorescence. Nuclear DNA was stained with DAPI. b) Percentage of cells with nucleoplasmic or nucleolar WRN before and after irradiation. c) WRN protein levels before and after irradiation in HEL cells analyzed by immunoblotting. d) Colocalization of WRN with DNA double strand break (DSB) repair foci after exposure of HEL cells to 10 Gy of irradiation. Immunofluorescence was performed 6 h after irradiation using antibodies against WRN (red), γ-H2AX (green), and ATMser1981-p (green). e) Clearance of γ-H2AX after irradiation was attenuated in WRN knockdown cells. Immunofluorescence staining was performed before and 0.5, 3, 6, and 12 h after irradiation. The percentage of cells with >10 γ-H2AX foci was statistically analyzed.
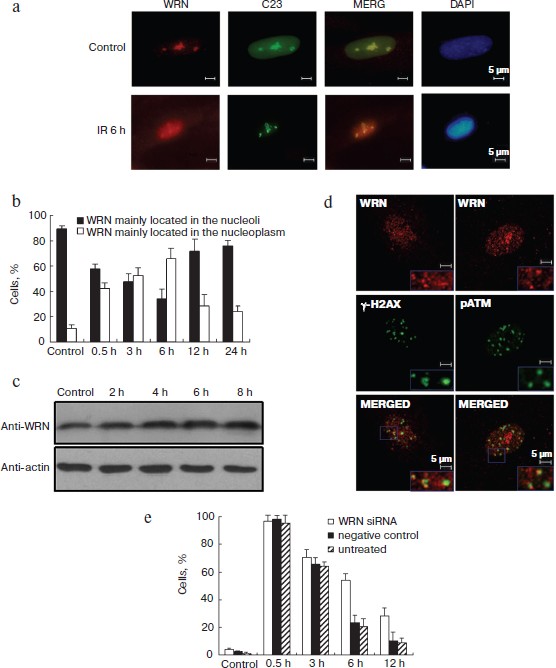
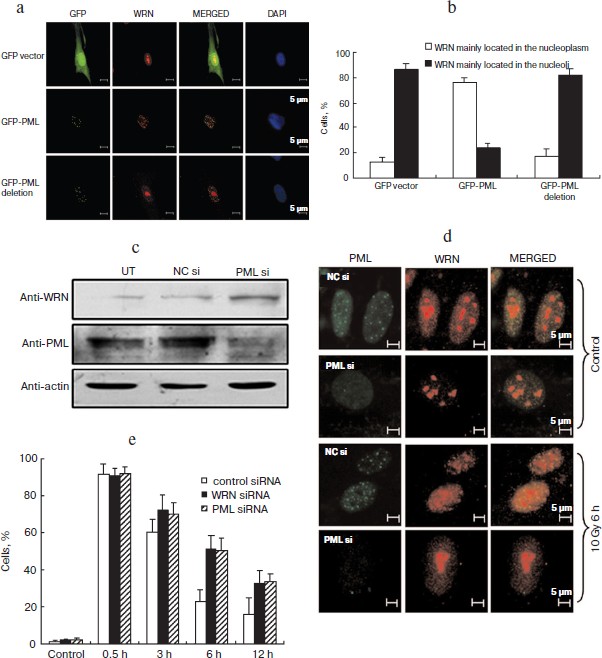
PML regulates irradiation-induced WRN relocation and DSB repair. It has been reported that ectopically expressed GFP-WRN localizes in the nucleoplasm in a speckled staining pattern and partly colocalizes with PML in UV and TSA treated U2OS cells [6, 7]. Considering the dynamic PML localization we observed in prior experiments (Fig. S1a in Supplement), we next examined the functional correlation between PML and WRN. As shown in Fig. 2a, top panel (see color insert), the nuclear localization of WRN in the pEGFP transfected HEL cells was similar to that in the untreated control cells (Fig. 1a). In pEGFP-PML IV transfected cells, WRN redistributed into the nucleoplasm (Figs. 2a, middle panel, and 2b), similar to cells exposed to irradiation.
To confirm the effect of PML on WRN localization after γ-irradiation, PML-siRNA was used. Down-regulation of PML was associated with a significant reduction in WRN nucleoplasmic localization. Distinct nucleoplasmic WRN foci were formed in untransfected control (UT) and negatively siRNA-transfected control (NC) cells following irradiation, but the majority of WRN in PML knockdown cells remained in nucleoli (Fig. 2, c and d). Furthermore, quantification of γ-H2AX foci showed that siPML and siWRN similarly delayed the repair of DSBs (Fig. 2e (color insert) and Fig. S4b (Supplement)). These results strongly support the hypothesis that PML is required for WRN translocation and accumulation at DSBs, and the translocation and accumulation are important in DSB repair in γ-irradiation-induced DNA damage.Fig. 2. PML regulates irradiation-induced WRN translocation and DSB repair. PML over-expression induced the nucleoplasmic translocation of WRN (a, b); down-regulation of PML perturbed WRN translocation to the nucleoplasm and DSB repair after irradiation (c, d, e). a) WRN was immunostained (red) in GFP, GFP-PML IV, or GFP-PML del 394-433 transfected HEL cells. Expression of GFP-PML IV but not GFP-PML del 394-433 mutants in HEL cells induced WRN relocalization from nucleoli to the nucleoplasm. b) Percentage of cells with nucleoplasmic or nucleolar WRN in PML IV or PML del 394-433 mutants over-expressed HEL cells and control cells. c) Knockdown of PML expression in HEL cells. Proteins were collected 48 h after siRNA transfection and analyzed by specific antibodies, as indicated. d) Relocalization of WRN at DSBs after irradiation was repressed by PML knockdown. Immunofluorescence was performed 6 h after irradiation using antibodies against WRN and PML. e) Clearance of γ-H2AX after irradiation was attenuated in PML or WRN knockdown cells. Immunofluorescence staining was performed and analyzed as in Fig. 1e.
WRN is physically associated with PML. To determine whether the γ-irradiation-induced endogenous WRN nucleoplasmic foci were colocalized with PML, immunofluorescence analysis of PML and WRN by confocal microscopy was performed before and after irradiation. The results shown in Fig. 3a (see color insert) demonstrate that only a small amount of endogenous WRN was localized in the nucleoplasm and overlapped with PML in unirradiated HEL cells; however, 6 h after irradiation, WRN was translocated to the nucleoplasm, and the colocalization of PML and WRN increased significantly.
To support the hypothesis that PML is associated with WRN in vivo, we performed co-immunoprecipitation using cell lysates from HEL cells. The results indicated that the anti-PML antibody co-immunoprecipitated endogenous PML and WRN (Fig. 3b). In addition, the GFP-tagged PML IV expression plasmid (pEGFP-PML) was transfected into A549 cells, and an analysis of anti-WRN immunoprecipitates demonstrated association between WRN and GFP-PML IV (data are not shown).Fig. 3. Interaction between WRN and PML. a) WRN partially colocalized with PML. Immunofluorescence staining was performed before and 6 h after irradiation using antibodies against PML (green) and WRN (red). b) WRN associates with PML. Lysates were immunoprecipitated with anti-PML antibody followed by immunoblotting using the indicated antibodies. c, d, e) The 394-433 aa domain of PML is important for PML–WRN interaction. c) The domain structure of full-length PML IV protein and the various PML IV truncation or deletion mutants. d) Co-immunoprecipitation was performed 24 h after pCMV-Myc-PML transfection. Myc antibodies were used for immunoprecipitation. The precipitates were analyzed by immunoblotting with WRN or Myc specific antibodies. e) EGFP–WRN and several Myc-tagged PML IV truncation mutants were over-expressed in A549 cells. Co-immunoprecipitation was performed with GFP antibody. The precipitates were analyzed by immunoblotting with GFP or Myc specific antibodies.
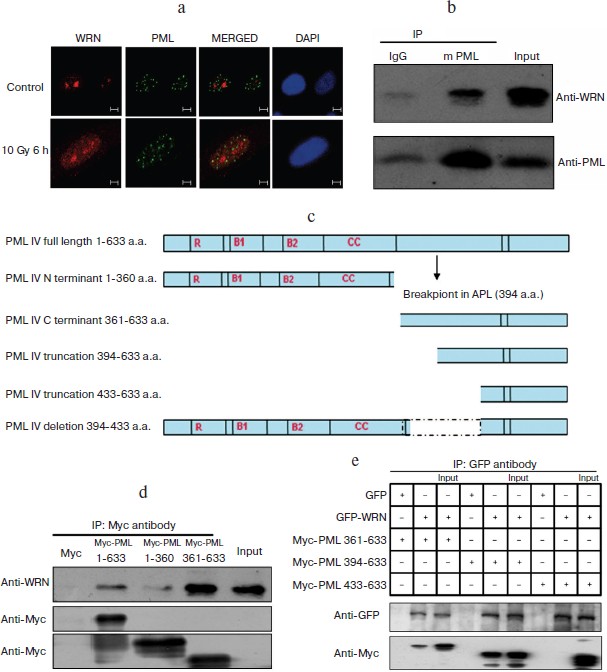
To define the regions of the PML protein involved in WRN binding, a Myc-tagged full-length PML IV and several PML IV truncation mutant expression DNAs were generated by PCR and ectopically expressed in A549 or MCF-7 cells (Fig. 3c). WRN was co-immunoprecipitated with Myc-tagged full-length PML IV (1-633 a.a.) or PML C-terminal truncation mutants Myc-PML IV (361-633 a.a.) and Myc-PML IV (394-633 a.a.). However, WRN could not be precipitated with Myc-PML IV (433-633) (Fig. 3, d and e), suggesting that amino acids 394 to 433 of PML are necessary for association with WRN.
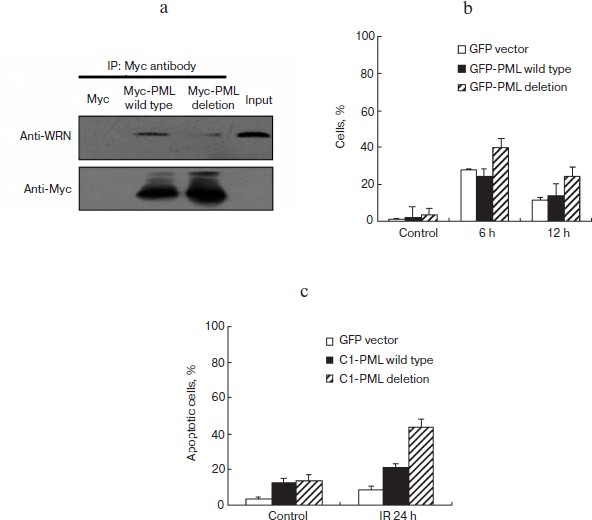
Interaction with PML is necessary for WRN translocation and DSB repair. To understand the functional significance of PML–WRN interaction further, GFP or the Myc-tagged PML IV deletion protein (PML del 394-433, which is depleted of the domain that confers WRN association) were ectopically expressed in HEL and MCF-7 cells. Compared with wild type PML IV, PML IV del 394-433 protein could not immunoprecipitate endogenous WRN (Fig. 4a; see color insert) and could not promote the transport of WRN from nucleoli to the nucleoplasm (Figs. 2a, bottom panel, and 2b).
In addition, the kinetics of phosphorylated histone H2AX (γ-H2AX) clearance after γ-irradiation showed that DSB repair appears to be delayed in GFP-PML del 394-433 transfected HEL cells as compared to GFP-PML IV transfected HEL cells (Fig. 4b).
Furthermore, quantitative analysis of apoptosis was performed in GFP, GFP-PML IV, and GFP-PML del 394-433 transfected HEL cells 24 h after irradiation. The GFP-PML del 394-433 transfected cells displayed higher sensitivity to irradiation-induced apoptosis compared to GFP-PML IV transfected cells (Fig. 4c). Similar results were obtained when we examined the DSB repair and radiosensitivity of GFP-PML IV and GFP-PML del 394-433 transfected MCF-7 cells (data not shown). The low capacity for DSB repair conferred by the 394-433 a.a. domain deletion in PML IV may contribute to the increased susceptibility to irradiation. Taken together, these data suggest that the 394-433 a.a. domain of PML IV is necessary for PML–WRN interaction, WRN nucleoplasmic translocation, and DSB repair in γ-irradiation-induced DNA damage response.
Fig. 4. The 394-433 a.a. domain of PML is important for PML–WRN interaction, WRN relocation, and DSB repair. a) The 394-433 a.a. domain of PML is important for PML–WRN interaction. Myc-tagged full-length PML and PML del (394-433) mutants were expressed in MCF-7 cells. Immunoprecipitations were performed by Myc specific antibodies. The precipitates were analyzed by Myc and WRN antibodies. b) Clearance of γ-H2AX after irradiation was delayed in GFP-PML del 394-433 expressed cells compared to pGFP-PML IV expressed HEL cells. Immunofluorescence staining was performed 6 and 12 h after 10 Gy-irradiation and analyzed by confocal microscopy. The percentage of cells with >10 γ-H2AX foci was calculated. c) GFP-PML del 394-433 transfected HEL cells were more sensitive to irradiation compared with pGFP-PML IV transfected HEL cells. Fluorescence flow cytometry analysis of cell apoptosis after irradiation was performed 24 h after irradiation. Propidium iodide was used as a marker for DNA content, and Annexin V was used as a marker for apoptotic cells.
DISCUSSION
Several proteins that participate in DSB signal transduction or repair form “repair foci”, also known as IRIF (ionizing radiation induced foci), at damage sites [12]. Ser139 phosphorylated histone H2AX (γ-H2AX) and ser1981 phosphorylated ATM (ATMser1981-p) are thought to be the most important and earliest components in these “repair foci” [13]. Bohr has reported that WRN was colocalized and associated with γ-H2AX following irradiation [3]. As H2AX may also be phosphorylated in the absence of direct DSB induction, e.g. with catalytic subunit of DNA-dependent protein kinase (DNA-PK) in replication stress [14, 15], the colocalization of ATMser1981-p and γ-H2AX with WRN was simultaneously analyzed to determine whether WRN is translocated to the site of DSBs induced by irradiation. ATMser1981-p activated by DSBs was localized in discrete foci, and other lesion-activated ATMser1981-p appeared diffusely scattered in the nucleoplasm [16]. Our results demonstrate that the WRN accumulated at the irradiation-induced DSB repair foci colocalized with ATMser1981-p and γ-H2AX. Together with the data that WRN knockdown cells displayed reduced DSB repair, these results suggested that WRN is relocalized to the DSB site to contribute to damage repair.
During DSB repair, the early smaller IRIFs subsequently aggregate into larger foci that are thought to represent unrepairable or slowly repaired DSBs [17]. In our study, the number of the cells with a high number of γ-H2AX foci (i.e. >10) began decreasing 3 h after irradiation. However, the cells with nucleoplasmic WRN foci increased progressively until 6 h after irradiation, showing that the increase of nucleoplasmic WRN foci coincided with the reduction of γ-H2AX foci. WRN was colocalized to larger γ-H2AX foci at a relatively late stage, suggesting that WRN may be involved in the repair of the more refractory lesions. In agreement with previous reports [18, 19], we observed that PML was partially colocalized with γ-H2AX and ATMser1981-p 6 h after irradiation (Fig. S3 in Supplement), raising the possibility that PML might be colocalized and function with WRN at DSBs at relatively late time points.
Blander et al. reported that ectopically expressed GFP-WRN forms bodies that partially colocalize with PML [6]. More detailed studies [7], where authors used ectopically expressed mRFP-WRN and EGFP-PML, showed that the surfaces of mRFP-WRN bodies were covered by a shell of EGFP-PML. The authors postulated that PML-covered mRFP-WRN bodies may be targeted for degradation. Here, we advanced this hypothesis by demonstrating an increase in endogenous WRN in PML knockdown HEL cells and a slight decrease in WRN in GFP-PML IV overexpressed cells. In addition, we show here that endogenous PML IV physically interacts with WRN and plays a crucial role in regulating the location of WRN after irradiation, and the 394-433 a.a. domain of PML IV was necessary for PML–WRN interaction, WRN nucleoplasmic translocation, and DSB repair. Thus, the relationship between WRN expression and location regulation that PML is involved in awaits further study.
PML NBs (promyelocytic leukemia nuclear bodies) have been reported to play a role in maintaining genome stability through interaction with and regulation of the location, posttranslational modification, and activity of many important factors (such as p53) involved in the DNA damage response. It has been reported that posttranslational modifications of WRN contribute to its distribution and enzymatic activity [2, 20, 21]. Proteins found to regulate posttranslational modification of WRN such as ATM, p300, Sirt1, and sumo-1, have been reported to localize with PML under certain circumstances [6, 7]. Our findings raise the possibility that PML NBs may also act as platforms for the posttranslational modification and functional regulation of WRN in γ-irradiation induced DNA damage responses. Furthermore, interactions of WRN with p53 or other associated proteins, such as NBS1, may also happen in PML NBs. It is of importance in the future to identify and characterize whether PML could regulate the posttranslational modification of WRN. It should be mentioned here that we were not able to acquire WRN-PML IV interaction by GST-pull down, so it is possible that the PML–WRN interaction needs other cofactors, or the interaction is dependent on some posttranslational modification of either of these two proteins that does not occur in E. coli.
Until now, at least seven PML isoforms have been reported [22]. We demonstrated here that PML IV can interact with WRN and regulate WRN translocation and DSB repair after irradiation through its 394-433 a.a. Interesting, the 394-433 a.a. domain of PML IV also exists in other PML isoforms (excluding the PML VII). Thus further studies are needed to evaluate whether other PML isoforms are also involved in WRN regulation.
Considering the effects of WRN in maintenance of genomic integrity, our findings provide new clues for understanding the function of PML as a cellular hub in DNA damage response.
We are grateful to Dr. Ying Luo and Dr. Xiao-fei Zheng for the HEL cells and their helpful advice.
This work was supported by grants from the Chinese National Key Program for Developing Basic Research (2007CB914604) and Chinese National Natural Sciences Foundation Projects (30600109, 30970683, and 30600162).
REFERENCES
1.Muftuoglu, M., Oshima, J., von Kobbe, C.,
Cheng, W. H., Leistritz, D. F., and Bohr, V. A. (2008) Hum.
Genet., 124, 369-377.
2.Sakamoto, S., Nishikawa, K., Heo, S. J.,
Goto, M., Furuichi, Y., and Shimamoto, A. (2001) Genes
Cells, 6, 421-430.
3.Cheng, W. H., Sakamoto, S., Fox, J. T., Komatsu,
K., Carney, J., and Bohr, V. A. (2005) FEBS Lett., 579,
1350-1356.
4.Lan, L., Nakajima, S., Komatsu, K., Nussenzweig,
A., Shimamoto, A., and Oshima, J. (2005) J. Cell Sci.,
118, 4153-4162.
5.Dellaire, G., and Bazett-Jones, D. P. (2004)
Bioessays, 26, 963-977.
6.Blander, G., Zalle, N., Daniely, Y., Taplick, J.,
Gray, M. D., and Oren, M. (2002) J. Biol. Chem., 277,
50934-50940.
7.Vaitiekunaite, R., Butkiewicz, D.,
Krzesniak, M., Przybylek, M., Gryc, A., Snietura, M.,
Benedyk, M., Harris, C. C., and Rusin, M. (2007) Mech. Ageing
Dev., 128, 650-661.
8.Bao-Lei, T., Zhu-Zhong, M., Yi, S., Jun-Jie,
Q., Yan, D., and Hua, L. (2006) J. Cell Biochem., 97,
561-571.
9.Bruno, S., Ghiotto, F., Fais, F., Fagioli, M.,
Luzi, L., and Pelicci, P. G. (2003) Blood, 101,
3514-3519.
10.Futami, K., Takagi, M., Shimamoto, A.,
Sugimoto, M., and Furuichi, Y. (2007) Biol. Pharm Bull.,
30, 1958-1961.
11.Kinner, A., Wu, W., Staudt, C., and Iliakis, G.
(2008) Nucleic Acids Res., 36, 5678-5694.
12.Gerashchenko, B. I., and Dynlacht, J. R. (2008)
Cytometry A, 75A, 245-252.
13.Tanaka, T., Huang, X., Halicka, H. D., Zhao, H.,
Traganos, F., and Albino, A. P. (2007) Cytometry A, 71,
648-661.
14.Kurose, A., Tanaka, T., Huang, X., Traganos, F.,
Dai, W., and Darzynkiewicz, Z. (2006) Cytometry A, 69,
212-221.
15.Burhans, W. C., and Weinberger, M. (2007)
Nucleic Acids Res., 35, 7545-7556.
16.Bakkenist, C. J., and Kastan, M. B. (2003)
Nature, 421, 499-506.
17.Mirzoeva, O. K., and Petrini, J. H. (2001)
Mol. Cell Biol., 21, 281-288.
18.Carbone, R., Pearson, M., Minucci, S., and
Pelicci, P. G. (2002) Oncogene, 21, 1633-1640.
19.Xu, Z. X., Timanova-Atanasova, A., Zhao, R. X.,
and Chang, K. S. (2003) Mol. Cell Biol., 23,
4247-4256.
20.Kahyo, T., Mostoslavsky, R., Goto, M., and
Setou, M. (2008) FEBS Lett., 582, 2479-2483.
21.Li, K., Casta, A., Wang, R., Lozada, E., Fan, W.,
and Kane, S. (2008) J. Biol. Chem., 283, 7590-7598.
22.Kirsten, J., Carol, S., and Paul, S. F. (2001)
Oncogene, 20, 7223-7233.
Supplementary Figures (MS Word)