In vivo Injected Mitochondria-Targeted Plastoquinone Antioxidant SkQR1 Prevents β-Amyloid-Induced Decay of Long-Term Potentiation in Rat Hippocampal Slices
N. A. Kapay1*, N. K. Isaev1,2,3*, E. V. Stelmashook1,3, O. V. Popova1, D. B. Zorov2,3, V. G. Skrebitsky1, and V. P. Skulachev2,3
1Department of Brain Research, Research Center of Neurology, Russian Academy of Medical Sciences, Pereulok Obukha 5, 105064 Moscow, Russia; E-mail: kapayn@gmail.com2Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: isaev@genebee.msu.ru
3Institute of Mitoengineering, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: info@skq-project.ru
* To whom correspondence should be addressed.
Received September 13, 2011; Revision received October 17, 2011
Addition of 200 nM β-amyloid 1-42 (Abeta) to a rat hippocampal slice impairs the induction of a long-term post-tetanic potentiation (LTP) of population spike (PS) in pyramidal neurons of the CA1 field of hippocampus. Intraperitoneal injection into the rat of the mitochondria-targeted plastoquinone derivative SkQR1 (1 µmol/kg of weight given 24 h before the slices were made) abolishes the deleterious effect of Abeta on LTP. These data demonstrate that SkQR1 therapy is able to compensate the Abeta-induced impairments of long-term synaptic plasticity in the hippocampus, which are the main cause of loss of memory and other cognitive functions associated with Alzheimer’s disease.
KEY WORDS: Alzheimer’s disease, β-amyloid, long-term memory, long-term potentiation, neurons, hippocampus, mitochondria-targeted antioxidants, SkQR1DOI: 10.1134/S0006297911120108
Abbreviations: Abeta, β-amyloid peptide 1-42; HFS, high frequency stimulation; LTP, long-term post-tetanic potentiation of hippocampal pathway; PS, population spike; ROS, reactive oxygen species; SkQR1, 10-(6′-plastoquinonyl)decylrhodamine 19.
Alzheimer’s disease is a widespread neurodegenerative pathology
characterized by pronounced dementia and loss of cognitive functions
and by development of extended degenerative impairments in the brain in
its later stages [1]. The β-amyloid peptide
(Abeta) is thought to be an essential factor in Alzheimer’s
disease pathogenesis. Submicromolar concentrations of this peptide are
known to impair synaptic transmission in glutamate synapses [2], whereas its micromolar concentrations cause
apoptotic-type neurodegeneration [3]. Mechanisms of
toxic effects of Abeta are not clear in detail. However, some data
suggest that a mitochondrial toxicity of Abeta is caused in particular
by stimulation of generation of reactive oxygen species (ROS) in
mitochondria [4-9]. It was
shown that Abeta, using the protein import system, enters the
mitochondrial matrix [10] where it interacts with
key mitochondrial enzymes including components controlling appearance
of the nonspecific permeability of the inner mitochondrial membrane [11]. The excessive generation of mitochondrial ROS
caused by Abeta with possible burst-like time course [12] could be crucial for the initiation by this
peptide of synaptic dysfunction and memory impairment in
Alzheimer’s disease. It was suggested that at least some
impairments could be prevented by mitochondria-targeted antioxidants
[13], and the present study has confirmed this
suggestion using a model of synaptic changes associated with learning
and memory (long-term potentiation of synaptic transmission in
hippocampus).
MATERIALS AND METHODS
The experiments were performed using hippocampal slices from young male Wistar rats (body weight 80-110 g). Freshly prepared slices were placed into a recording chamber and perfused with modified Ringer solution of the following composition (mM): NaCl, 124; KCl, 3; CaCl2, 2.5; MgSO4, 2.5; Na2HPO4, 1.25; NaHCO3, 26; D-glucose, 10, constantly saturated with Carbogen (95% O2 + 5% CO2) at 29-30°C. The electrical activity was recorded starting from 1.5-2 h after the slices were prepared. A focal response caused by stimulation of the radial layer by solitary rectangular impulses (duration 0.1 msec, interval 1/15 sec) was recorded in the pyramidal layer of the CA1 field using a glass microelectrode filled with 1.5 M NaCl (resistance, 2-5 mΩ). The impulse power was chosen to provide the amplitude of the peak component of the response representing the summary spike response of the pyramidal neuron population (pop-spike, PS) to be about a half of its maximum value. LTP of PS was induced by high-frequency stimulation (HFS, 100 Hz, 1 sec) by the same electrodes and at the same power of the stimulus as in control. Each slice was subjected to only one HFS. Fifteen minutes before the HFS, the flow was redirected from a second reservoir with a solution containing Abeta. The reverse switching was performed 5 min after the HFS. Changes in the reactivity of the pyramidal neurons were assessed by changes in the PS amplitude relative to its mean value determined by a 15-min background recording (the period before the change of the flow).
Concentrated aqueous solutions of Abeta (Sigma-Aldrich, USA) were stored as frozen aliquots. The stored Abeta solution was diluted with the perfusion medium immediately before use. SkQR1 (1 µmol/kg) was intraperitoneally injected into rats 24 h before preparing the slices.
Statistical analysis of mean values and mean errors (M ± m) were done using the nonparametric Mann–Whitney test and Student’s test.
The experimental protocols on treatment of animals were performed according to 86/609/EEC Regulations of the European Society Council for the use of animals in experiments and approved by the Moscow State University Ethics Commission.
RESULTS
In our experiments, the standard HFS (100 Hz, 1 sec) of Shaffer’s collaterals caused LTP of PS in the CA1 hippocampal field resulting 30 min after tetanization in an increase in the PS amplitude to 145.9 ± 7.8% (n = 6) in the control rats (figure, panel (a)). In hippocampal slices from rats that received 1 µmol SkQR1 per kg weight 24 h before isolation, the LTP amplitude was slightly higher than in the control, i.e. 175.4 ± 17.1% (n = 6) (figure, panels (b) and (d)).
Perfusion of slices with a solution containing 200 nM Abeta did not cause a significant change in the basal PS but impaired the ability of pyramidal neurons of the hippocampal field CA1 to form LTP after the HFS (figure, panels (a) and (c)). The mean value of the PS amplitude upon 15-min perfusion of the slices with the Abeta-containing solution (before the HFS) was 73.0 ± 13.4%, and 30 min after the HFS it became 57.1 ± 10.8% (n = 5). The difference of the latter value from the control is significant (P < 0.01; figure, panel (d)).
SkQR1 abolishes the inhibitory effect of β-amyloid peptide 1-42 (Abeta) on LTP in the hippocampus. The arrow indicates the HFS start and the box shows duration of treatment of the slices with 200 nM Abeta. a) Population spike (PS) in the hippocampal slices prepared from the control animals and perfused with solution without Abeta (n = 6); b) PS in the hippocampal slices prepared from the animals pretreated with SkQR1 (1 µmol/kg) and perfused with Abeta-free solution (n = 6); c) PS in hippocampal slices treated with 200 nM Abeta, obtained from the control animals not treated with SkQR1 (n = 5) (circles) or from the animals in vivo pretreated with SkQR1 (1 µmol/kg) (n = 6) (rhombs); d) mean amplitudes of population spike recorded 30 min after HFS. * P < 0.01 as compared to Abeta; ** P < 0.01 as compared to control
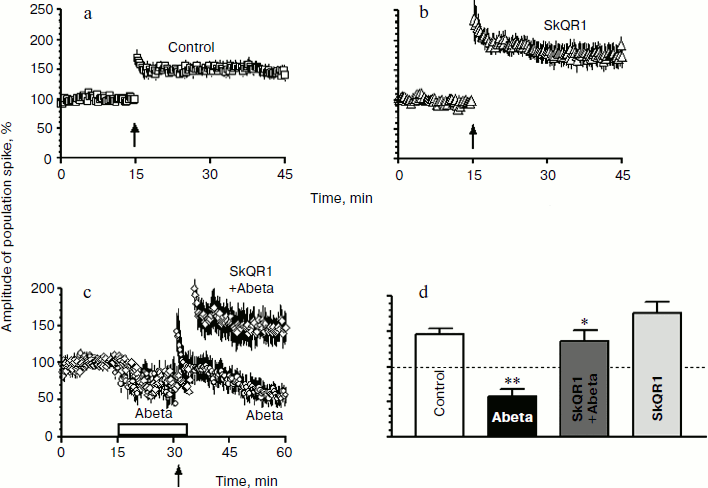
Abeta-treated hippocampal slices from the rats pretreated with SkQR1 showed response indicating that SkQR1 abolishes the inhibitory effect of Abeta on the LTP induction. The mean value of the PS amplitude in the SkQR1 + Abeta samples was 74.1 ± 5.5 and 135.7 ± 16.2% before and after HFS, respectively (n = 6). The latter value was significantly higher than the corresponding one obtained in the Abeta-treated slices from the rats that did not obtain SkQR1 (P < 0.01) (figure, panels (c) and (d)).
Thus, we have shown for the first time that the injection into animals of the mitochondria-targeted antioxidant SkQR1 abolishes the inhibitory effect of Abeta on LTP in the hippocampus.
DISCUSSION
It was demonstrated earlier that very low (nM) concentrations of mitochondria-targeted plastoquinone derivatives SkQ1 and SkQR1 behave as highly effective antioxidants in aqueous solutions, lipid micelles, liposomes, isolated mitochondria, and cell cultures [14]. These penetrating cations can be used for treatment of a number of ROS-associated age-related diseases in humans and animals, including “dry eye” syndrome, cardiac arrhythmia, myocardial and kidney infarction, and brain stroke [15-17]. We have shown that a single intraperitoneal injection to rats of SkQR1 (0.5-2 µmol per kg body weight) decreases the infarction zone caused by compression ischemia or occlusion of the middle carotid artery [15, 17]. In the latter case, the decrease in the damaged zone correlated with the attenuation of the neurological deficit [17]. Earlier, positive anti-ischemic effect of another mitochondria-targeted antioxidant (MitoQ) was demonstrated in the model of heart ischemia [18], but this compound was inefficient in the case of brain ischemia [19].
Alzheimer’s disease is a common age-related disease that seems to be mediated by mitochondrial ROS. This disease is characterized by a progressing loss of memory and other cognitive functions. The production of Abeta from its protein precursor is a key event in the development of this disease [4]. The accumulation of Abeta in cells leads to synaptic dysfunction and memory impairments [2, 20, 21]. The Abeta-triggered pathological cascade in the initial phase may be a result of mitochondrial oxidative stress that in terminal phase gives rise to expression of the proapoptotic enzyme GSK3β [22]. It seems reasonable to suggest that a pathological increase in the ROS level and hence memory impairments associated with this disease could be prevented by mitochondria-targeted antioxidants [13].
The electrical response of hippocampal slices, namely the long-term potentiation of synaptic transmission in the hippocampal pathway (the Schaffer collaterals-CA1 field), can be used as a model of synaptic changes associated with learning and memory [23]. Using this model, we have shown that 15 min treatment with 200 nM Abeta impairs LTP induction in the rat hippocampus. The intraperitoneal injection into animals of the mitochondria-targeted plastoquinone derivative, antioxidant SkQR1 (1 µmol per kg body weight) 24 h before the slice preparation abolishes the inhibitory effect of Abeta on the LTP in the slices. Note that concurrently with us Ma et al. [5] studied the effect of another mitochondria-targeted antioxidant MitoQ [5] using it to prevent the effect of Abeta on LTP. They supplemented the antioxidant in vitro, i.e. to the hippocampal slices, whereas in our experiments SkQR1 was injected in vivo. Moreover, Ma et al. [5] observed that the addition of Abeta to hippocampal slices induced hyperproduction of mitochondrial ROS that could be prevented by addition of MitoQ to the perfusion medium. These findings demonstrate the efficiency of mitochondria-targeted antioxidants as promising drugs for treatment the Alzheimer’s disease.
Mitochondria-targeted antioxidants can apparently correct Abeta-caused impairments in glutamatergic neuronal transmission that underlie dementia and the loss of other cognitive functions in Alzheimer’s disease. In this case SkQR1 may be more effective than MitoQ due to its much higher antioxidant activity [24]. Moreover, SkQR1 was shown to be capable not only directly to lower the level of mitochondrial ROS but also to elevate the ischemic tolerance through an increase in concentration of erythropoietin in vivo [25]. The latter effect results in a decrease in activity of the proapoptotic enzyme GSK3β involved in the neuronal Abeta toxic cascade [13, 22].
REFERENCES
1.Terry, R. D., Masliah, E., Salmon, D. P., Butters,
N., Deteresa, R., Hill, R., Hansen, L. A., and Katzman, R. (1991)
Ann. Neurol., 30, 572-580.
2.Selkoe, D. J. (2002) Science, 298,
789-791.
3.Kimura, M., Akasofu, S., Ogura, H., and Sawada, K.
(2005) Brain Res., 1047, 72-84.
4.Pagani, L., and Eckert, A. (2011) Int. J.
Alzheimer’s Dis., DOI: 10.4061/2011/925050.
5.Ma, T., Hoeffer, C. A., Wong, H., Massaad, C. A.,
Zhou, P., Iadecola, C., Murphy, M. P., Pautler, R. G., and Klann, E.
(2011) J. Neurosci., 31, 5589-5595.
6.Tillement, L., Lecanu, L., and Papadopoulos, V.
(2011) Mitochondrion, 11, 13-21.
7.Cardoso, S. M., Swerdlow, R. H., and Oliveira, C.
R. (2002) Brain Res., 931, 117-125.
8.Yan, S. D., Xiong, W. C., and Stern, D. M. (2006)
J. Alzheimer’s Dis., 9, 127-137.
9.Newington, J. T., Pitts, A., Chien, A., Arseneault,
R., Schubert, D., and Cumming, R. C. (2011) PLoS One, 6,
e19191.
10.Hansson Petersen, C. A., Alikhani, N., Behbahani,
H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., Leinonen, V., Ito, A.,
Winblad, B., Glaser, E., and Ankarcrona, M. (2008) Proc. Natl. Acad.
Sci. USA, 105, 13145-13150.
11.Singh, P., Suman, S., Chandna, S., and Das, T. K.
(2009) Bioinformation, 3, 440-445.
12.Zorov, D. B., Filburn, C. R., Klotz, L. O.,
Zweier, J. L., and Sollott, S. J. (2000) J. Exp. Med.,
192, 1001-1014.
13.Skulachev, V. P. (2011) J. Alzheimer’s
Dis., 28, DOI: 103233/JAD-2011-111391.
14.Antonenko, Y. N., Avetisyan, A. V., Bakeeva, L.
E., Chernyak, B. V., Chertkov, V. A., Domnina, L. V., Ivanova, O. Y.,
Izyumov, D. S., Khailova, L. S., Korshunova, G. A., Lyamzaev, K. G.,
Muntyan, M. S., Nepryakhina, O. K., Pashkovskaya, A. A., Pletyushkina,
O. Y., Pustovidko, A. V., Roginsky, V. A., Rokitskaya, T. I., Ruuge, E.
K., Saprunova, V. B., Severina, I. I., Simonyan, R. A., Skulachev, I.
V., Skulachev, M. V., Sumbatyan, N. V., Sviryaeva, I. V., Tashlitsky,
V. N., Vasil’ev, J. M., Vyssokikh, M. Y., Yaguzhinsky, L. S.,
Zamyatnin, A. A., and Skulachev, V. P. (2008) Biochemistry
(Moscow), 73, 1273-1287.
15.Bakeeva, L. E., Barskov, I. V., Egorov, M. V.,
Isaev, N. K., Kapelko, V. I., Kazachenko, A. V., Kirpatovsky, V. I.,
Kozlovsky, S. V., Lakomkin, V. L., Levina, S. V., Pisarenko, O. I.,
Plotnikov, E. Y., Saprunova, V. B., Serebryakova, L. I., Skulachev, M.
V., Stelmashuk, E. V., Studneva, I. M., Tskitishvili, O. V.,
Vasil’eva, A. K., Victorov, I. V., Zorov, D. B., and Skulachev,
V. P. (2008) Biochemistry (Moscow), 73, 1288-1299.
16.Skulachev, V. P., Anisimov, V. N., Antonenko, Y.
N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., Filenko, O. F.,
Kalinina, N. I., Kapelko, V. I., Kolosova, N. G., Kopnin, B. P.,
Korshunova, G. A., Lichinitser, M. R., Obukhova, L. A., Pasyukova, E.
G., Pisarenko, O. I., Roginsky, V. A., Ruuge, E. K., Senin, I. I.,
Severina, I. I., Skulachev, M. V., Spivak, I. M., Tashlitsky, V. N.,
Tkachuk, V. A., Vyssokikh, M. Y., Yaguzhinsky, L. S., and Zorov, D. B.
(2009) Biochim. Biophys. Acta, 1787, 437-461.
17.Plotnikov, E. Y., Silachev, D. N., Chupyrkina, A.
A., Danshina, M. I., Yankauskas, S. S., Morosanova, M. A., Stelmashuk,
E. V., Vasil’eva, A. K., Goryacheva, E. S., Pirogov, Y. A.,
Isaev, N. K., and Zorov, D. B. (2010) Biochemistry (Moscow),
75, 145-150.
18.Adlam, V. J., Harrison, J. C., Porteous, C. M.,
James, A. M., Smith, R. A., Murphy, M. P., and Sammut, I. A. (2005)
FASEB J., 19, 1088-1095.
19.Hobbs, C. E., Murphy, M. P., Smith, R. A., and
Oorschot, D. E. (2008) Pediatr. Int., 50, 481-488.
20.Oddo, S., Caccamo, A., Shepherd, J. D., Murphy,
M. P., Golde, T. E., Kayed, R., Metherate, R., Mattson, M. P., Akbari,
Y., and LaFerla, F. M. (2003) Neuron, 39, 409-421.
21.Haas, C., and Selkoe, D. J. (2007) Nat. Rev.
Mol Cell. Biol., 8, 101-112.
22.Lloret, A., Badia, M. C., Giraldo, E., Ermak, G.,
Alonso, M. D., Pallardo, F. V., Davies, K. J. A., and Vina, J.
(2011) J. Alzheimer’s Dis., DOI:
10.3233/JAD-2011-110890.
23.Malenka, R. C., and Nicoll, R. A. (1999)
Science, 285, 1870-1874.
24.Skulachev, M. V., Antonenko, Y. N., Anisimov, V.
N., Chernyak, B. V., Cherepanov, D. A., Chistyakov, V. A., Egorov, M.
V., Kolosova, N. G., Korshunova, G. A., Lyamzaev, K. G., Plotnikov, E.
Y., Roginsky, V. A., Savchenko, A. Y., Severina, I. I., Severin, F. F.,
Shkurat, T. P., Tashlitsky, V. N., Shidlovsky, K. M., Vyssokikh, M. Y.,
Zamyatnin, A. A., Jr., Zorov, D. B., and Skulachev, V. P. (2011)
Curr. Drug Targets, 12, 800-826.
25.Plotnikov, E. Y., Chupyrkina, A. A., Jankauskas,
S. S., Pevzner, I. B., Silachev, D. N., Skulachev, V. P., and Zorov, D.
B. (2011) Biochim. Biophys. Acta, 1812, 77-86.