A New Viral Vector Exploiting RNA Polymerase I-Mediated Transcription
T. V. Komarova1,2, A. M. Schwartz2, A. A. Makarov2, and Yu. L. Dorokhov1,2*
1Vavilov Institute of General Genetics, Russian Academy of Sciences, ul. Gubkina 3, 119991 Moscow, Russia; fax: (499) 132-89622Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia; fax: (495) 939-3181; E-mail: dorokhov@genebee.msu.su
* To whom correspondence should be addressed.
Received January 18, 2012; Revision received January 31, 2012
We have developed a new viral vector system exploiting RNA-polymerase I transcription. The vector is based on the crucifer-infecting tobacco mosaic virus (crTMV) cDNA inserted into the rRNA transcriptional cassette (promoter and terminator). To visualize reproduction of the vector, the coat protein gene was replaced with the gene encoding green fluorescent protein (GFP) resulting in a PrrRNA-crTMV-GFP construct. Our results showed that agroinjection of Nicotiana benthamiana leaves with this vector results in GFP production from uncapped crTMV-GFP RNA because RNA polymerase I mediates synthesis of rRNA lacking a cap. Coexpression of the crTMV 122 kDa capping protein gene and the silencing suppressor encoded by the tomato bushy stunt virus p19 gene stimulated virus-directed GFP production more than 100-fold. We conclude that the Pol I promoter can be used to drive transcription in a transient expression system.
KEY WORDS: tobacco mosaic virus, viral vector, crTMV, agroinjection, protein superproduction, RNA polymerase IDOI: 10.1134/S0006297912050148
Abbreviations: crTMV, crucifer-infecting TMV; IC, intermediate construct; IRES, internal ribosome entry site; TBSV, tomato bushy stunt virus; TMV, tobacco mosaic virus.
Three different RNA polymerases (Pols), designated Pol I, II, and III,
catalyze basic transcription [1]. All Pol
II-transcribed RNAs have a 5′-cap structure that is important for
pre-mRNA processing, mRNA export, translation, and quality control [2-4]. Pol I is involved in the
high-level synthesis of the non-capped single-transcript rRNA precursor
(pre-rRNA), which is processed into 28S (25S in plants), 5.8S, and 18S
rRNAs [5-7].
Tobacco mosaic virus (TMV) replicates in the cytoplasm, and its genome is a single-stranded sense RNA (~6.5 kb) with an m7GpppG cap structure on its 5′ end [8-10]. One of the TMV replicase components, the 126 kDa protein, performs the viral genomic RNA capping that is essential for recruiting translation initiation factors [11, 12] and for anchoring RNA to the actin/endoplasmic reticulum (ER) network [13]. Capping of viral RNA occurs through the guanylyltransferase activity of the vital 126 kDa capping enzyme [14, 15].
Agroinfection or Agrobacterium-delivered TMV vector insertion leads to virus infection of almost all leaf cells [16]. In the transient expression system, TMV-based viral vector is delivered to the cell nucleus with the help of A. tumefaciens T-DNA. Then, Pol II-synthesized viral capped genomic RNA is exported from the nucleus into the cytoplasm where it is translated to produce its own capping enzyme replicase complex. However, under natural conditions, plant RNA viruses replicate in the cytoplasm and are not adapted to the nuclear splicing machinery, which recognizes and removes cryptic introns from viral RNA, leading to the degradation of that RNA. Recently, to increase the level of infectious transcripts, the expensive method of TMV cDNA modification was developed. This method involves the removal of putative cryptic splice sites and the insertion of multiple plant introns [16].
We have created a system exploiting Pol I-directed transcription that does not require nuclear splicing. Previously, the Pol IrRNA promoter was used in hybrid constructions for expressing noncoding shRNAs [17, 18] or negative-strand animal viral RNA molecules [19-24]. The Pol I (rRNA) promoter has an advantage in RNA production compared to Pol II. It was shown that Pol I-synthesized RNA molecules do not compete for nucleocytoplasmic export with Pol II-synthesized RNA [17]. This finding is important in cases of co-agroinjection of Pol I- and Pol II-based vectors.
We constructed a plasmid, designated Pol IrRNA-crTMV-GFP, containing the full-length cDNA of the crucifer-infecting TMV strain (crTMV) [25] and the GFP gene instead of the CP gene and showed that the amount of genomic viral RNA exported to the cytoplasm was sufficient for the initiation of crTMV replication and GFP production. Although the level of GFP accumulation was low, co-expression of the crTMV 122 kDa capping protein gene and the silencing suppressor tomato bushy stunt virus (TBSV) p19 gene stimulated virus-directed GFP production more than 100-fold.
MATERIALS AND METHODS
Plasmid and vectors. To construct the Pol I promoter-based vectors, we used the pBor2 plasmid [26] that contains the Brassica oleracea rRNA gene promoter sequence from –518 to +106. The Pol I-based infectious copy of crTMV-GFP was created via several intermediate constructs (IC).
IC1. Using overlap PCR, fragment 1, which contained the 114 bp 3′ proximal to the Pol I-promoter fused to the 112 bp 5′ proximal to the crTMV genome, was obtained. Plasmids pBor2 and pICH4351 [27] were used as templates for PCR. The 5′-region of the Pol I-promoter from pBor2 flanked by and digested with HindIII/SphI (fragment 2) and fragment 1 digested with SphI/MunI were inserted into pSP72 (Promega, USA) digested with HindIII/MunI, resulting in intermediate construct 1 (IC1). Primers used for PCR were as follows: PolI-SphI-Dir1 (CCTTATGAAGCATGCAAAAAATCAGATTC), PolI-Rev (ATATAGACAAAACACTTTTTTTCAG), 4351-Dir2 (AAAAAGTGTTTTGTCTATATaGTTTTAGTTTTATTGCAACAACAAC), and 4351-MunI-Rev2 (GCTTGGAGAGTTTGCATGTC).
Overlap PCR for the analog, IC1*, was performed using pICH17388 [16] as a template; IC1* contained two A. thaliana introns in the 5′-part of the crTMV genome.
IC2. For the Pol IrRNA-5′IRES-crTMV-GFP construct, IRESCP,148 was amplified from crTMV-CP-GFP [28]. Fragment 3 containing the 114 bp 3′ proximal to the Pol I-promoter fused to IRESCP,148 and the 112 bp 5′ proximal to the crTMV genome was obtained by overlap PCR using the following primers: PolI-SphI-Dir1, PolI-Rev, 4351-MunI-Rev2, p1-IRES_D1 (GTGTTTTGTCTATATAATTCGTCGATTCGGTTG), IRES-omega_R1 (GCAATAAAACTAAAACTTTCTTCTTTCAAATTAAACGAATC), and IRES-omega_D1 (GAAAGAAGAAAGTTTTAGTTTTATTGCAACAAC). To obtain IC2, fragment 3 was digested with SphI/MunI and inserted into pSP72 containing the 5′ part of Pol IrRNA (fragment 2).
IC3. For the Pol IrRNA-IRES3′-crTMV-GFP construct, IRESCP,148 was amplified from crTMV-CP-GFP [28]. Fragment 4 containing the 114 3′ proximal nucleotides of the Pol I-promoter fused to the 68 bp 5′ proximal to the crTMV genome (omega sequence) followed by IRESCP,148 and the first 44 bp of the crTMV RdRp ORF was obtained by overlap PCR using the following primers: PolI-SphI-Dir1, PolI-Rev, 4351-MunI-Rev2, 4351-Dir2, Omega-IRES_R1 (GCTGCAACCGAATCGACGAATTGTTGTTGTTGTTTGTATTTTG), IRES_D2 (AATTCGTCGATTCGGTTGCAGCAT), IRES_R3 (ATTTTCTTCTTTCAAATTAAACGAATC), and IRES_D4 (CGTTTAATTTGAAAGAAGAAAATGGCACAATTTCAAC). To obtain IC3, fragment 4 was digested with SphI/MunI and inserted into pSP72 containing fragment 2.
IC4. To obtain a fragment containing the Saccharomyces cerevisiae Pol I-terminator of transcription, the corresponding oligonucleotides were annealed pairwise, extended with Klenow fragment, and then amplified. The following yeast-specific overlapping primers were used: yT1-BHI-dir (TTTGGATCCCTCGAGGGTACCGCATGCAGATCTTAGA), yT1-rev (TAAAATTTATAGAGACTTGTTCAGTCTAAGATCTGCA), yT2-dir (CAAGTCTCTATAAATTTTATTTGTCTTAAGAATTCTA), and yT2-rev (ACTGAGCTCTTTTTACCCGGATCATAGAATTCTTAAG). This fragment 5, which was flanked by BamHI/SacI sites, was digested and inserted into pCambia1300 (Cambia, Australia) with a fragment of pICH4351 [27] flanked by PstI/BamHI sites and containing the 3′ proximal part of the replicase, GFP, and the 3′ UTR. The resulting plasmid was IC4.
In the final cloning step, fragments flanked by HindIII/MunI were obtained from IC1, IC2, and IC3. The construct crTMV-CP-GFP was digested with MunI/PstI to produce the fragment containing the part of the crTMV genome from 87 to 3670 nt. The fragments from IC1, IC2, and IC3 were ligated into IC4 digested with HindIII/PstI, resulting in the Pol I-crTMV-GFP viral vector (in the case of the IC1 fragment), Pol IrRNA-5′IRES-crTMV-GFP (for the IC2 fragment), or Pol IrRNA-IRES3′-crTMV-GFP (for the IC3 fragment). IC1* was used for the intron-containing vector, Pol IrRNA-crTMV(intr)-GFP.
To obtain the Pol IIAct2-122 kDa construct, PCR was performed with the primers 122 kDa-HIII-fwd (CCCGGTTGTGGAAAAACGAAGG) and 122 kDa-BsrGI-rev (TTTTGTACAATTGGACCCCCGCTTCAAC). The resulting fragment containing Pol IIAct2 and the 122 kDa coding sequence was digested with HindIII/BsrGI and inserted into crTMV-CP-GFP using the same sites.
The KpnI/ScaI fragment from pICH4351 (containing the promoter and the MT domain) was inserted into the crTMV 122 kDa construct digested with KpnI/SmaI resulting in a plasmid containing the 122 kDa methyl-transferase (MT) domain and named Pol IIAct2-122 kDa MT.
All stages of cloning were performed using Fermentas (Lithuania) enzymes.
Agroinjection. A 500 µl sample of an overnight A. tumefaciens (GV-3101 strain) culture was sedimented at 2300g for 5 min, and the pellet was resuspended in a solution containing 10 mM MES (pH 5.5) and 10 mM MgCl2 to a final OD600 = 0.1. When a combination of different strains was used, aliquots of each strain with OD600 = 0.2-0.3 were mixed. Leaves of greenhouse grown Nicotiana benthamiana plants were injected with this suspension using a syringe without a needle [16, 27].
SDS-PAGE and Western blot analysis. Total soluble protein was extracted from agroinfiltrated N. benthamiana leaves. Samples were ground with Celite in phosphate buffered saline (PBS). Crude leaf extracts were resolved on 12% SDS-polyacrylamide gel using Laemmli’s buffer system. For Western blot analysis, fractionated proteins were transferred to Hybond-P PVDF membranes (GE Healthcare, UK), blocked with 5% skim milk (Fluka, Germany) in TBS (Tris-buffered saline supplemented with 0.1% Tween-20), and probed with GFP-specific HRP-conjugated antibodies (Rockland Immunochemicals, USA) diluted 1 : 15,000 in TBS. The Western blot was developed with ECL detection reagent (GE Healthcare). Densitometric analysis was performed with Scion Image software (Scion Corp.).
GFP imaging. GFP fluorescence in inoculated leaves was monitored by illumination with a handheld UV source (DESAGA). Fluorescence microscopy was performed using a Zeiss Axiovert 200M microscope (Zeiss, Germany) with two sets of filters, BP470/40 (excitation) and BP525/50 (emission).
GFP fluorescence measurement. Leaf tissue (50 mg) was homogenized in 200 μl of GFP-extraction buffer (200 mM NaCl, 10 mM Tris-HCl, pH 8.0). After centrifugation, the volume of supernatant was adjusted to 1 ml. GFP fluorescence was measured using a QUANTECH fluorimeter (Barnstead International, USA) equipped with filters NB390 (excitation) and NB520 (emission).
RNA extraction. Total RNA was isolated from plant tissue using TriReagent (MRC, USA) according to the manufacturer’s protocol.
Reverse transcription and real-time qPCR analysis. Total cDNA was obtained by annealing 2 μg of denatured total RNA with 0.1 μg of random hexamers and 2 pmol of GFP-RT primer followed by incubation with 200 units of Superscript II reverse-transcriptase (Invitrogen, USA) according to the manufacturer’s protocol. Real-time qPCR was carried out using the iCycler iQ real-time qPCR detection system (Bio-Rad, USA). For the detection of target genes, the Eva Green master mix (Syntol, Russia) was used according to the manufacturer’s instructions. Each sample was run in triplicate, and a non-template control was added to each run. Target gene mRNA levels were corrected by comparison with RNA levels for the reference gene (18S rRNA). The following oligonucleotides were used: GFP-F1_RT (GCAGAAGAACGGCATCAAG), GFP-R1_RT (GCTCAGGTAGTGGTTGTCG), GFP-RT (CTCGTCCATGCCGTGAGTGAT), 18S_F1 (ACGGCTACCACATCCAAG), and 18S_R1 (ACTCATTCCAATTACCAGACTC).
RESULTS AND DISCUSSION
Construction of Pol IrRNA-crTMV-GFP vector. The crTMV strain was used to construct the infectious cDNA vectors (Fig. 1a) under control of the B. oleracea rRNA promoter (Pol IrRNA) and the Saccharomyces cerevisiae terminator. The crTMV genomic RNA is 6312 nt in length, has a cap on its 5′-end, and contains four open reading frames (ORFs). Two components of the replicase complex, 122 and 178 kDa, are encoded by the 5′-proximal ORFs and translated from the viral genomic RNA. The movement protein (MP) and the coat protein (CP) genes are expressed from subgenomic RNAs (sgRNA) that are transcribed from subgenomic promoters in the negative strand of genomic RNA.
To determine if a Pol I promoter can direct the synthesis of crTMV genomic RNA, we constructed a plasmid containing the full-length cDNA of crTMV with GFP instead of the CP gene, designated Pol IrRNA-crTMV-GFP (Fig. 1a, upper). To indicate the cells in which viral replication was initiated, the vector was constructed so that GFP was expressed from the sgRNA made by the viral replicase in infected cells. We did not observe visible fluorescence sites (see, for example, Fig. 3c, site 1), but microscopic study revealed a few clusters of fluorescent cells (Fig. 1b). However, the level of GFP expression was low compared to an analogous vector, Pol IIAct2-crTMV-GFP [28], in which GFP expression was under the control of the Pol II actin promoter (Arabidopsis thaliana Act2 promoter).Fig. 1. The Pol IrRNA-promoter-based crTMV vector directs GFP synthesis in N. benthamiana leaves. a) Schematic of the Pol IrRNA-crTMV-GFP (upper) and intron-containing Pol IrRNA-crTMV(intr)-GFP (lower) vectors. Pol I, B. oleracea rRNA promoter; RdRp, RNA-dependent RNA polymerase gene; MP, movement protein gene; GFP, green fluorescent protein gene; term, yeast Pol I transcription terminator. b) Fluorescence microscopy photograph of a group of cells under UV light 5 days after inoculation with Pol IrRNA-crTMV-GFP. c) Western blot analysis of GFP in the total soluble protein from N. benthamiana leaves after agroinjection with Pol IrRNA-crTMV-GFP (lanes 1 and 2), Pol IrRNA-crTMV(intr)-GFP (lanes 3 and 4), and Pol IIAct2-crTMV-GFP (lane 5). The lower panel is the protein loading control stained with amido-black. d) Relative quantity of GFP mRNA, determined by real-time qPCR in leaf areas after agroinjection with Pol IrRNA-or Pol IIAct2-directed TMV-GFP with (+) or without (w/o) the TBSV p19 silencing suppressor.
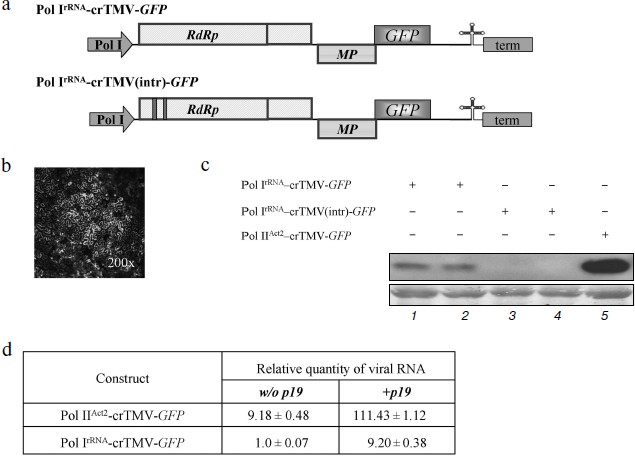
Thus, the Pol IrRNA-crTMV-GFP vector directed GFP synthesis in agroinjected leaves but with low efficiency. This conclusion was confirmed by Western blot analysis of total soluble protein from the agroinjected leaves developed with GFP-specific antibodies (Fig. 1c, lanes 1 and 2).
To verify that transcription of Pol IrRNA-crTMV-GFP was performed by Pol I and not Pol II, we constructed a vector (Pol IrRNA-crTMV(intr)-GFP) containing two A. thaliana introns in the 5′-proximal part of the ORF encoding a 122 kDa protein (Fig. 1a, lower). Normally, when transcription is performed by Pol II, these introns are excised during splicing, producing the mRNA from which the fully functional proteins of the replicase complex (122 and 178 kDa) are synthesized. However, if the introns are not removed correctly, the 122 kDa ORF is interrupted by stop codons, no functional 122 and 178 kDa proteins can be synthesized, and no viral vector replication can occur. In contrast, the Pol I export route does not include splicing, so intron insertion into crTMV genomic RNA abolishes either its RNA export or synthesis of the replicase complex. No fluorescence was detected in leaf areas agroinjected with Pol IrRNA-crTMV(intr)-GFP, and Western blot analysis revealed no GFP production from that viral vector (Fig. 1c, lanes 3 and 4).
The assessment of viral RNA accumulation by real-time PCR showed that the amount of GFP mRNA from Pol IrRNA-crTMV-GFP is approximately 9-fold lower than from Pol IIAct2-crTMV-GFP (Fig. 1d, column 1). Our results also showed that co-delivery of the plasmid 35S-p19 [27] encoding the silencing suppressor, p19, of tomato bushy stunt virus (TBSV) resulted in an approximately 9-fold increase in viral vector RNA accumulation (Fig. 1d, column 2). To explain this result, we suggested that because RNA synthesized by Pol I does not carry any “shield” (a cap, for example) on the 5′-end, these RNAs could be the target of different cellular exonucleases. Moreover, any viral RNA entering the plant cell activates the host defense system and switches on virus-induced gene silencing (VIGS) that leads to degradation of foreign RNA. Viral RNA protection and suppression of VIGS, especially during the first round of replicase synthesis, may facilitate the subsequent start of replication and thus the increase in GFP production.
We conclude that Pol I-directed transcription may result in the appearance of crTMV RNA in the cytoplasm and that TBSV p19 stabilizes Pol I-directed viral progeny RNA.
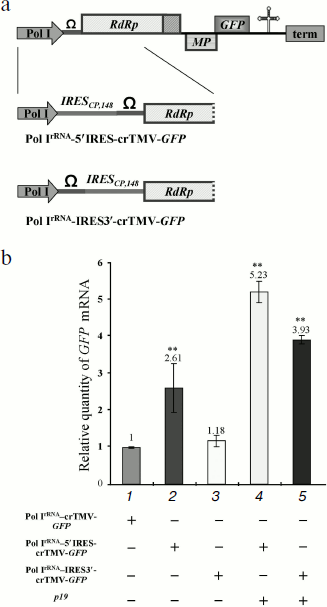
IRESCP,148 enhances Pol I-directed crTMV reproduction. The main drawbacks of a Pol I promoter-based system for positive RNA viruses are the inability to make capped, sense viral transcripts to protect the RNA from cellular exonucleases and the inability to recruit translation initiation factors. We suggested that insertion of an internal ribosome entry site (IRES) could partially compensate for the lack of a cap. To test that hypothesis, we exploited the crTMV IRESCP,148 [29, 30]. In the case of the crTMV-based viral vector, it was not clear whether the IRES should be inserted at the 5′-end of the genomic RNA or between the 5′-untranslated region (omega, Ω) and the replicase coding sequence. Two IRES-containing constructs were created (Fig. 2a). In the first construct, Pol IrRNA-IRES3′-crTMV-GFP, the IRESCP,148 was inserted between omega and the replicase gene because the 5′-untranslated region should not be important for ribosome internal entry. The infectivity of that viral vector seems to be slightly higher than that of the basic construct, Pol IrRNA-crTMV-GFP, measured by real-time qPCR (Fig. 2b, graph bar 3). The negligible increase in infectivity may be due to the IRES in this position impairing replicase binding to the cis-element(s) required for replication. Inserting the IRESCP,148 just upstream of omega, so as not to interrupt the natural coding sequence of the viral genomic RNA, increased infectivity of the resulting construct, Pol IrRNA-5′IRES-crTMV-GFP, 2.5-fold (Fig. 2b, graph bar 2). Silencing suppression after p19 co-injection increased the accumulation of RNA directed by both of the IRES-containing constructs more than 2-fold (Fig. 2b, graph bars 4 and 5). Thus, the IRESCP,148 is able to partially compensate for the lack of the viral RNA cap.
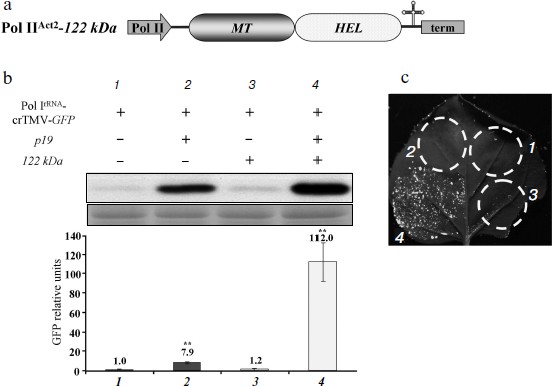
The MT domain of the crTMV 122 kDa gene enhances Pol I-directed crTMV reproduction. It is well known that the methyltransferase activity of the replicase protein 126 kDa TMV U1 is responsible for capping at the first step of viral genomic RNA replication [14, 15]. We assessed whether the crTMV capping enzyme could stimulate viral reproduction in trans by protecting viral genomic RNA from exonucleases by adding a cap-structure at its 5′-end. To do so, we constructed a plasmid containing the crTMV 122 kDa gene under control of the Pol IIAct2-promoter (Fig. 3a). Joint agroinjection of the Pol IrRNA-crTMV-GFP vector and Pol IIAct2-122 kDa may result in only a slight (15-20%) increase in GFP production, measured by densitometry of the band on Western blots (Fig. 3b, graph bar 3). Western blot analysis and densitometry demonstrated more than a 100-fold stimulation of GFP expression from Pol IrRNA-crTMV-GFP when the 122 kDa gene was accompanied by the p19 gene (Fig. 3b, graph bar 4). In contrast, p19 alone increased GFP expression only approximately 8-fold (Fig. 3b, graph bar 2). These results were also confirmed by the visual detection of GFP expression in leaf sections (Fig. 3c). Thus, p19-mediated protection from the plant defense systems of both Pol IrRNA-directed crTMV-GFP and Pol II-directed 122 kDa RNAs strongly increased the effect achieved by 122 kDa co-expression. Taking into account that Pol IrRNA-crTMV-GFP-mediated GFP production level in the presence of p19 is 9-fold lower than Pol IIAct2-crTMV-GFP-mediated (Fig. 1d), we claim that in combination with the capping enzyme and p19 (Fig. 3b, No. 4) Pol IrRNA-crTMV-GFP provides the comparable with Pol IIAct2-crTMV-GFP level of GFP expression.Fig. 2. The crTMV IRESCP,148 is able to partially compensate for the lack of the viral RNA cap. a) Schematic representation of the Pol IrRNA-promoter-based crTMV vectors with the IRESCP,148 in two positions, upstream (Pol IrRNA-5′IRES-crTMV-GFP) and downstream (Pol IrRNA-IRES3′-crTMV-GFP) of the omega (Ω) sequence. Pol I, rRNA promoter; RdRp, RNA-dependent RNA polymerase gene; MP, movement protein gene; GFP, green fluorescent protein gene; term, yeast Pol I transcription terminator. b) Relative quantity of GFP mRNA, determined by real-time qPCR in leaf areas after agroinjection with the IRESCP,148-containing Pol IrRNA-based vectors. ** P < 0.05 (unpaired two-tailed Student’s t-test).
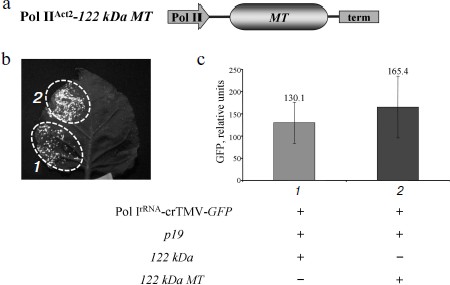
To show that the 122 kDa enhancer effect on Pol IrRNA-crTMV-GFP is determined by its N-terminal methyltransferase (MT) domain, which is responsible for capping, we created an A. thaliana Act2 promoter-based construct, Pol IIAct2-122 kDa MT, encoding the MT domain of the crTMV replicase (Fig. 4a). The truncated 122 kDa gene with the MT domain retained its enhancer properties (Fig. 4b) as confirmed by fluorescence quantification of GFP expression (Fig. 4c). These data demonstrate that the MT domain of the crTMV 122 kDa protein is responsible for enhancing Pol I-directed crTMV reproduction.Fig. 3. The 122 kDa capping enzyme and TBSV p19 silencing suppressor stimulate Pol IrRNA-directed crTMV-GFP reproduction. a) Schematic of the vector PrAct2-122 kDa. Pol II, A. thaliana Act2 promoter; MT, methyltransferase domain; HEL, helicase domain; term, nos-terminator of transcription. b) Western blot (upper) of GFP in the total soluble protein from N. benthamiana leaves 5 days after agroinjection and the relative density of GFP bands of five independent experiments (bottom). The lower panel is the protein loading control stained with amido-black. ** P < 0.05 (Student’s t-test). c) Visualization of GFP expression under UV light 5 days after co-agroinjection with GFP-encoding crTMV constructs.
Collectively, our results show that a simple GFP-encoding crTMV vector based on an rRNA transcriptional cassette (promoter and terminator) is able to produce genomic viral RNA sufficient for crTMV replication despite the lack of RNA capping. Pol I transcripts resulted in GFP production that became significant after viral RNA capping and stabilization. Our transient system provided GFP yields comparable to those of Pol II promoter-based vectors.Fig. 4. The crTMV 122 kDa methyltransferase domain is responsible for enhancement of Pol IrRNA-directed crTMV-GFP reproduction. a) Schematic of the vector, Pol IIAct2-122 kDa MT. Act2, A. thaliana Act2 promoter; MT, methyltransferase domain sequence of 122 kDa encoding gene; term, nos-terminator. b) GFP visualization in N. benthamiana leaves 5 days after agroinjection. c) GFP fluorimetric analysis. GFP relative units are the result of five independent experiments. P > 0.05 (Student’s t-test).
The authors are grateful to Dr. C. S. Pikaard and to Dr. Y. Gleba for generously providing plasmids.
This work was supported by the Russian Foundation for Basic Research grant No. 11-04-01152-a (TVK) and Federal Agency for Science and Innovations grants No. 16.512.11.2268 (YLD) and 16.512.11.2279 (YLD).
REFERENCES
1.Cramer, P., Armache, K. J., Baumli, S., Benkert,
S., Brueckner, F., et al. (2008) Annu. Rev. Biophys., 37,
337-352.
2.Furuichi, Y., and Shatkin, A. J. (2000) Adv.
Virus Res., 55, 135-184.
3.Gu, M., and Lima, C. D. (2005) Curr. Opin.
Struct. Biol., 15, 99-106.
4.Lewis, J. D., and Izaurralde, E. (1997) Eur. J.
Biochem., 247, 461-469.
5.Granneman, S., and Baserga, S. J. (2005) Curr.
Opin. Cell. Biol., 17, 281-287.
6.Kuhn, C. D., Geiger, S. R., Baumli, S., Gartmann,
M., Gerber, J., et al. (2007) Cell, 131, 1260-1272.
7.Moss, T., Langlois, F., Gagnon-Kugler, T., and
Stefanovsky, V. (2007) Cell. Mol. Life Sci., 64,
29-49.
8.Efimov, V. A., Chakhmakcheva, O. G., Archdeacon,
J., Fernandez, J. M., Fedorkin, O. N., et al. (2001) Nucleic Acids
Res., 29, 4751-4759.
9.Goelet, P., Lomonossoff, G. P., Butler, G. J. P.,
Akam, M. E., Gait, M. J., et al. (1982) Proc. Natl. Acad. Sci.
USA, 79, 5818-5822.
10.Zimmern, D. (1975) Nucleic Acids Res.,
2, 1189-1201.
11.Pelham, H. R. B. (1978) Nature,
272, 469-471.
12.Wilson, T. M. A. (1984) Virology,
137, 255-265.
13.Christensen, N., Tilsner, J., Bell, K., Hammann,
P., Parton, R., et al. (2009) Traffic, 10, 536-551.
14.Merits, A., Kettunen, R., Makinen, K., Lampio,
A., Auvinen, P., et al. (1999) FEBS Lett., 455,
45-48.
15.Dunigan, D. D., and Zaitlin, M. (1990) J.
Biol. Chem., 265, 7779-7786.
16.Marillonnet, S., Thoeringer, C., Kandzia, R.,
Klimyuk, V., and Gleba, Y. (2005) Nat. Biotechnol., 23,
718-723.
17.Komarova, T. V., Schwartz, A. M., Frolova, O. Y.,
Zvereva, A. S., Gleba, Y. Y., et al. (2010) Plant Mol. Biol.,
74, 591-603.
18.Brenz Verca, M. S., Weber, P., Mayer, C., Graf,
C., Refojo, D., et al. (2007) Nucleic Acids Res., 35,
e10.
19.Neumann, G., and Kawaoka, Y. (2002) Virus
Res., 82, 153-158.
20.Freiberg, A., Dolores, L. K., Enterlein, S., and
Flick, R. (2008) Virology, 370, 33-44.
21.Groseth, A., Feldmann, H., Theriault, S.,
Mehmetoglu, G., and Flick, R. (2005) J. Virol., 79,
4425-4433.
22.Billecocq, A., Gauliard, N., Le May, N., Elliott,
R. M., Flick, R., et al. (2008) Virology, 378,
377-384.
23.Flick, R., and Pettersson, R. F. (2001) J.
Virol., 75, 1643-1655.
24.Habjan, M., Penski, N., Spiegel, M., and Weber,
F. (2008) J. Gen. Virol., 89, 2157-2566.
25.Dorokhov, Yu. L., Ivanov, P. A., Novikov, V. K.,
Agranovsky, A. A., Morozov, S. Y., et al. (1994) FEBS Lett.,
350, 5-8.
26.Doelling, J. H., and Pikaard, C. S. (1996)
Nucleic Acids Res., 24, 4725-4732.
27.Marillonnet, S., Giritch, A., Gils, M., Kandzia,
R., Klimyuk, V., et al. (2004) Proc. Natl. Acad. Sci. USA,
101, 6852-6857.
28.Dorokhov, Y. L., Ivanov, P. A., Komarova, T. V.,
Skulachev, M. V., and Atabekov, J. G. (2006) J. Gen. Virol.,
87, 2693-2697.
29.Ivanov, P. A., Karpova, O. V., Skulachev, M. V.,
Tomashevskaya, O. L., Rodionova, N. P., et al. (1997) Virology,
232, 32-43.
30.Dorokhov, Y. L., Skulachev, M. V., Ivanov, P. A.,
Zvereva, S. D., Tjulkina, L. G., et al. (2002) Proc. Natl. Acad.
Sci. USA, 99, 5301-5306.