REVIEW: Mitochondrial Production of Reactive Oxygen Species
V. G. Grivennikova* and A. D. Vinogradov
Department of Biochemistry, Biological Faculty, Lomonosov Moscow State University, 119991 Moscow, Russia; E-mail: vgrivennikova@mail.ru; adv@biochem.bio.msu.su* To whom correspondence should be addressed.
Received June 18, 2013
Numerous biochemical studies are aimed at elucidating the sources and mechanisms of formation of reactive oxygen species (ROS) because they are involved in cellular, organ-, and tissue-specific physiology. Mitochondria along with other cellular organelles of eukaryotes contribute significantly to ROS formation and utilization. This review is a critical account of the mitochondrial ROS production and methods for their registration. The physiological and pathophysiological significance of the mitochondrially produced ROS are discussed.
KEY WORDS: oxidoreductases, respiratory chain, ROS production, mitochondriaDOI: 10.1134/S0006297913130087
Abbreviations: Amplex Red, 10-acetyl-3,7-dihydrophenoxazine; DLDH, dihydrolipoamide dehydrogenase; DODH, dihydroorotate dehydrogenase; ETF, electron-transferring flavoprotein; GSSG and GSH, oxidized and reduced forms of glutathione, respectively; MAO, monoamine oxidase; mGPDH, mitochondrial α-glycerophosphate dehydrogenase; NOX, NAD(P)H oxidase; O2•- and O2•H, superoxide anion and its protonated form; •OH, hydroxyl radical; OGDHc, α-oxoglutarate dehydrogenase complex; PDHc, pyruvate dehydrogenase complex; ROS, reactive oxygen species; SMP, submitochondrial particles; SOD, superoxide dismutase; Δp, proton electrochemical potential difference.
In the resting state, an adult man consumes about 10 mmol of oxygen
per minute and expires an almost equivalent amount of carbon
dioxide [1]. These numbers are increased up to
10-fold during moderate physical exercise such as walking on grass at
the rate of about 10 km/h. Simple calculations demonstrate an intensity
of oxidative metabolism: if oxidation of glucose to carbon dioxide and
water is tightly coupled with oxidative phosphorylation, and oxygen is
reduced exclusively by the mitochondrial cytochrome oxidase, resting
state respiration results in daily turnover (breakdown and resynthesis)
of about 40 kg of ATP! More than 90% of the oxygen consumed by mammals
is reduced to water by the mitochondrial cytochrome oxidase. Only a
small part is converted to partially reduced products (superoxide,
hydrogen peroxide, hydroxyl radical) conventionally called ROS
(reactive oxygen species). Since the late fifties of the previous
century, steadily increasing attention is paid to the participation of
these species in biochemistry and physiology. The most significant
steps in biochemistry of ROS are depicted in Table 1 in chronological order. The data reported in the
literature on mitochondrial ROS production, its dependence on metabolic
state, and their quantitative characteristics are countless and often
controversial. This can be illustrated by citations from several
publications such as: “The mitochondrial electron-transport chain
is the main source of ROS during normal metabolism” [16]. “The mitochondrial respiratory chain
constitutes the main intracellular source of ROS in most tissues”
[17]. “Are mitochondria a permanent source
of reactive oxygen species?” [18].
“There is no evidence that mitochondria are the main source of
reactive oxygen species in mammalian cells” [19]. It should also be noted that mitochondria from
various tissues are different in their relative and specific activities
of the enzymes participating in ROS metabolism and also in local oxygen
availability for terminal oxidation (liver, kidney, heart, vessel
endothelia, and lung).
In this review, we will focus mostly on ROS production by heart mitochondria. This is (i) because of our own experience in the field and (ii) because intact coupled heart mitochondria are easily available, as well as a number of simpler well-defined preparations derived therefrom.
We will discuss some properties of oxygen as an oxidizer. Basic information on the mitochondrial respiratory chain, the major cellular oxygen consumer, will be provided. The short description of other than respiratory chain component mitochondrial enzymes capable of ROS production is the subject of next section. Several notes concerning physiological and pathophysiological significance of mitochondrial ROS production will be made in the final part of this review.
Table 1. Landmarks in biochemistry of
ROS*

Note: The papers published up to 1976 are included in the table. During
the following years the number of publications on the subject have been
increasing exponentially. However, they do not report any basic new
concepts, but only expand and provide deeper understanding the ideas
that were proposed by the pioneers in the field. A reader interested in
“signaling” function of ROS [12, 13], their non-mitochondrial sources [14], and current state of the enzymology of
superoxide dismutases [15] is addressed to the
respective references.
CHEMISTRY OF OXYGEN REDUCTION
Oxygen is a strong oxidant. The meaning of the widely used term “strong oxidant” seems worth discussing as it is relevant to the subject of this review. Aerobic energetics of all organisms is supported by the free energy change in oxidoreduction between intracellular reductants (averaged redox potential is about –320 mV) and oxidant (dissolved oxygen redox potential in the reaction of water formation at neutral pH of about +800 mV). The meaning of the expression oxygen is a “strong” oxidant is that the reaction:
O2 + 4 ē + 4 H+ → 2 H2O (1)
is thermodynamically irreversible if electrons are provided by the cellular reducing components (mainly NADH). On the other hand, oxygen is a “poor” oxidant. Its electronic structure (two unpaired electrons with the same spins on different π-orbitals) does not allow accepting simultaneously two electrons with antiparallel spins from most organic compounds. Kinetic inertness of oxygen is the reason for chemical stability of many organic molecules in the environment containing 21% gaseous oxygen. Spin restriction in the reaction of oxidation of organic molecules by oxygen and thermodynamic irreversibility of these reactions are evidently the reasons why evolution has chosen oxygen as the major oxidant for aerobic life (fitness of oxygen [20]). Evolution has solved the kinetic inertness of oxygen by creating enzymes, oxidases that catalyze its reduction to water (cytochrome oxidases, reaction (1)) or to hydrogen peroxide (other oxidases, reaction (2)):
O2 + 2 ē + 2 H+ → H2O2. (2)
The spin restriction is avoided by these enzymes in such a way that they catalyze reaction (1) or (2) with aid of one-electron cofactors-donors, such as transition metal ions (iron, manganese, copper, molybdenum) and/or organic molecules capable of relatively stable free radical state (flavins, quinones). In contrast to the kinetically inert molecular oxygen, its intermediately reduced forms (ROS) are highly reactive, and some of them, i.e. hydroxyl radical, can oxidize nonenzymatically proteins, lipids, nucleic acids, and low molecular mass metabolites. Remarkably, most oxidases catalyze step-by-step reduction of oxygen to water or hydrogen peroxide with no release of partially reduced potentially dangerous oxygen species (see [21] and references therein). Only a small part of oxygen consumed is reduced to hydrogen peroxide (reaction (2)), which further serves as the oxidant in peroxidase reactions:
H2O2 + 2 ē + 2 H+ → 2 H2O (3)
or the reductant and oxidant in the dismutation reaction:
H2O2 + H2O2 → 2 H2O + O2. (4)
The presence of catalytically active transition metal ions and/or organic molecules capable of one-electron reactions as the cofactors in a number of oxidoreductases or oxygen carriers (myoglobin, hemoglobin) provide the possibility for one-electron reduction of oxygen to superoxide radical anion (pKa 4.7):
Direct evidence for enzymatic formation of superoxide radical was obtained in 1969 (xanthine oxidase activity of modified xanthine dehydrogenase, EC 1.17.3.2) [22]. Since then “xanthine oxidase” is most widely used as the source of superoxide in biochemical practice.
In aqueous solutions at pH > 5 superoxide is unstable and dismutates non-enzymatically:
2 O2•- + 2 H+ → O2 + H2O2 (6)
in the second-order reaction (rate constant of about 100 M–1⋅sec–1) [23]. In 1969, McCord and Fridovich showed that the red cell protein previously called erythrocuprein catalyzes reaction (6) at rate and substrate affinity (Km) similar to those known for other oxidoreductases [5]. The enzyme contains copper and zinc as cofactors. Later an isoenzyme containing manganese was found in prokaryotes [6].
One-electron reduction of hydrogen peroxide (the third electron on the way of four-electron step-by-step reduction of oxygen to water) results in formation of highly reactive (thermodynamically and kinetically) hydroxyl radical:
H2O2 + 1 ē → •OH + OH–. (7)
Superoxide radical itself can serve as a one-electron donor for hydrogen peroxide reduction in the reaction catalyzed by transition metals (Fe, Cu):
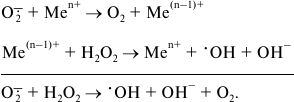 (8)
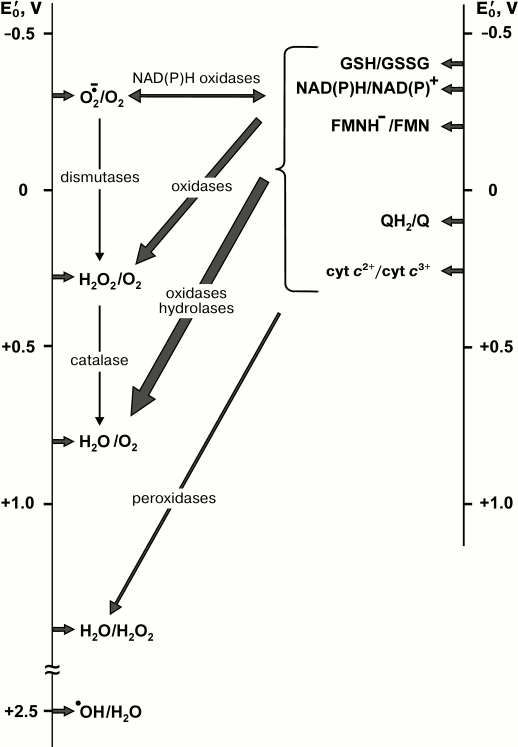
(8)Hydrogen peroxide in the presence of transition metal ions is widely used in chemistry as a strong oxidant (Fenton reagent). It should be noted that only hydroxyl radical is a highly “active” oxygen specie, but because superoxide and hydrogen peroxide participate in its formation (reactions (5)-(8)) all three partially reduced oxygen species are usually called “reactive”. Some oxygen-dependent enzymatic reactions and the standard redox potentials of their substrate/product pairs are schematically depicted in Fig. 1.
Fig. 1. Diagram of the major biochemical reactions of oxygen. Vertical lines are the scales of the standard redox potentials relative to the hydrogen electrode at pH 7.0. Arrows on left scale indicate the redox potentials of oxygen species, those on right scale correspond to the potentials of some substrates-electron donors. For the sake of simplicity, the potentials of the substrates of other catalyzed reactions such as mono-/diamino-, amino acid-, hexose oxidases, and many substrates of hydroxylases are not shown. For this information a reader is addressed to the classical book by Clark [24]. The values indicated on left scale are taken from reviews [20, 24-26].
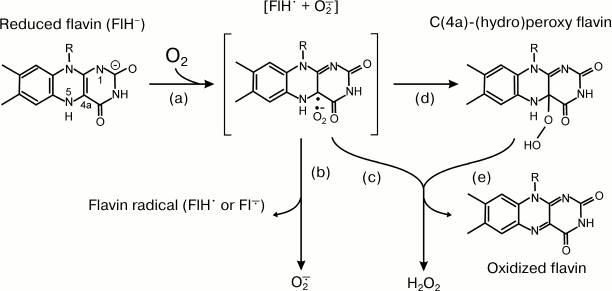
A number of oxidoreductases, which do not use oxygen as the substrate or product under aerobic conditions, are capable of production or utilization of ROS as side products (substrates). Most of them are flavoproteins, and the chemistry of ROS formation by reduced flavin is given as an example. Flavins (FMN and FAD) are able to form free radicals upon oxidoreduction. Many enzymes adopt this property for their electron transferase activity when they catalyze the oxidation of two-electron donors (most organic substrates) by one-electron acceptors (iron-sulfur clusters, cytochromes). Reduced protein-bound or free flavins are oxidized by oxygen, and depending on particular protein environment O2•- or H2O2 or both are formed. Figure 2 depicts the simplified scheme of the reaction between flavin and oxygen [27]. The reduced flavin (FlH–) donates one electron to oxygen, and the radical pair (FlH•⋅O2•-, step a) is formed. Homolytic cleavage (step b) results in the formation of superoxide and flavin radical, whereas heterolytic cleavage produces hydrogen peroxide and oxidized flavin either directly (step c) or via formation of (C(4a)-(hydro)peroxy intermediate (steps d and e).
Fig. 2. Reactions of reduced flavin with oxygen (adapted from [27]). See text for explanation.
Other reactions resulting in ROS formation include other one-electron redox components such as iron-sulfur clusters, cytochrome hemes, and ubisemiquinones.
HEART MITOCHONDRIA AND THEIR RESPIRATORY CHAIN
The constant contraction/relaxation of heart muscle requires considerable energy produced by mitochondria during aerobic oxidative phosphorylation. Mitochondria, making up 25-30% of myocardial mass, are located close to myofibrils, and well-developed intramitochondrial contacts form a reticulum [28, 29]. Fatty acids, glucose, and lactate are the major metabolic fuel for the heart, although pyruvate and ketone bodies supplied by blood are also substrates for oxidation [30, 31]. The structure of heart mitochondria is the same as in other tissues: they are vesicles surrounded by two closed membranes, outer and inner. The latter is folded, forming well-developed cristae, and the volume of the matrix surrounded by the inner membrane is small. The outer membrane separates mitochondria and cytosol and contains several enzymes and so-called porins, which provide osmotically neutral transport of metabolites of molecular mass <5 kDa into the intermembrane space. The main energy-producing machinery is located in the inner membrane. The respiratory chain consists of four components: respiratory complexes I (NADH:ubiquinone reductase), II (succinate:ubiquinone reductase), III (ubiquinol:cytochrome c reductase), and IV (cytochrome c:oxygen oxidoreductase, cytochrome oxidase). The fifth component of the oxidative phosphorylation system is ATP synthase (Fo⋅F1-ATPase). Electron transfer from NADH and succinate to oxygen results in formation of water, and energy released is accumulated as phosphoryl group transfer potential of ATP. The inner membrane contains many translocases, which catalyze highly selective transport of ions and neutral organic molecules in and out of the matrix. Mitochondria contain a number of other oxidoreductases operating with ubiquinone as the electron acceptor. “Soluble” enzymes operating in the matrix (Krebs cycle, β-oxidation of fatty acids, etc.) provide the reducing equivalents (NAD(P)H) for further oxidation by the respiratory chain.
PREPARATIONS USED FOR ASSAY OF MITOCHONDRIAL ROS
PRODUCTION
Preparations
A number of enzymatically active preparations are used in studies of mitochondrial ROS production: intact and permeabilized mitochondria, submitochondrial particles (SMP), purified components of the respiratory chain, and other oxidoreductases.
Intact mitochondria. In intact mitochondria, the formation of ROS by the membrane-bound enzymes and by the matrix-located proteins proceeds in their natural environment. The major advantage of intact mitochondria is that they are tightly coupled, different physiologically relevant states can be investigated such as resting state (no added ADP, slow respiration, state 4 according to Chance’s nomenclature [32]) or active state (in the presence of added ADP, rapid respiration, state 3 [32]). The major shortcoming in the use of intact mitochondria is the extreme complexity of the system. Consider a reaction that produces superoxide or hydrogen peroxide during NADH oxidation in the mitochondrial matrix. To measure overall ROS production, the substrate of NAD+-dependent dehydrogenase, i.e. malate for malate dehydrogenase located in matrix, is to be added. The equilibrium of the malate dehydrogenase reaction is strongly shifted towards oxaloacetate reduction, and added glutamate is required to trap oxaloacetate in a transaminase reaction. Alternatively, pyruvate can be added to trap oxaloacetate by acetyl-CoA and subsequent citrate formation. The rate of overall ROS formation would be a complex function of: (i) malate, glutamate, and aspartate (or pyruvate and citrate) transport; (ii) malate dehydrogenase and transaminase (or pyruvate dehydrogenase complex, PDHc, and citrate synthase) activities; (iii) ROS-producing enzyme activity; (iv) activities of the intramitochondrial ROS-utilizing enzymes (antioxidant defense system including matrix-located and intramembrane-located SODs, glutathione peroxidase, catalase, peroxyredoxins, glutaredoxins); and (v) transport of hydrogen peroxide across two membranes. Needless to say, it would be extremely difficult if not impossible to interpret what is the actual target of an effector that influences an experimentally observed rate of ROS production.
The inner mitochondrial membrane is non-permeable for superoxide anion [33, 34]. It is generally assumed that hydrogen peroxide freely penetrates the inner membrane. However, we have shown that this assumption is questionable: oxygen formation by catalase from hydrogen peroxide externally added to intact mitochondria was strongly stimulated by the pore-forming antibiotic alamethicin [35].
The use of alamethicin or other agents for permeabilization partially avoids transport limitations. Under certain conditions, alamethicin provides unlimited transfer of low molecular mass compounds to and from the mitochondrial matrix, leaving the residence of matrix-located proteins unaltered [36, 37]. On the other hand, permeabilization of the inner membrane results in a complete collapse of proton-motive force (Δp), a key parameter controlling mitochondrial metabolism.
Isolated purified enzymes. The advantage of isolated enzymes with no contaminating components is obvious. However, possible modifications of the membrane-bound proteins during their preparative procedure caused by ultrasonic treatment and by detergents cannot be excluded. Also, the effects of the membrane potential on their ROS-producing activity can only be studied after reconstitution into proteoliposomes.
Submitochondrial particles. SMP is the choice for studies on ROS generation and other activities catalyzed by the enzymes located in the inner mitochondrial membranes. Inside-out coupled SMP catalyze oxidation of NADH and succinate with respiratory control index (ratio of oxygen consumption at state 3 and 4) of about 8 and 3, respectively [38]. The substrate-binding active sites of the membrane-bound oxidoreductases exposed to matrix in intact mitochondria are directly accessible for added compounds in inside-out SMP. Also, SMP are almost completely devoid of the soluble matrix-located protein including the components of antioxidant defense. Substrate oxidation or ATP hydrolysis by SMP results in Δp formation, thus allowing studies on simulation of different physiological states (state 3 and 4 and their transition).
Methods of ROS Detection
Assay of superoxide. Superoxide detection is not simple even when it is produced by purified enzymes. The O2•- radical is not stable, and a number of redox active compounds that are rapidly and specifically oxidized or reduced by superoxide followed by determination of the products using optical or EPR methods are applied. The reduction of cytochrome c, epinephrine or dihydroethidine oxidation, and specific reactions with spin traps are used [39]. None of these reagents show absolute specificity for superoxide, and many oxidoreductases directly react with them. This necessitates calculations of the SOD-sensitive reaction, which should be a significant fraction of the overall reaction. Circumstantial indications of superoxide production are also widely used. For example, the iron-sulfur cluster of aconitase, the enzyme catalyzing interconversion of citrate to isocitrate, is sensitive to oxidation, and its catalytic activity is irreversibly destroyed by superoxide. The loss of aconitase activity is widely used as a criterion for superoxide-producing activity of intact mitochondria [39, 40]. Another way to assay superoxide production is to measure hydrogen peroxide that is formed in the presence of SOD [39, 41].
Assay of hydrogen peroxide. Rapid and specific high-affinity binding of hydrogen peroxide to heme-containing peroxidases results in the formation of a spectrally detectable complex, so-called compound I, and this reaction can be used for direct quantitative determination of H2O2 [41]. Other assay procedures based on the specific hydrogen peroxide–peroxidase interaction have been developed. They utilize peroxidation of a number of fluorescent or light-absorbing compounds-donors, such as scopoletin, diacetyldichlorofluorescein, p-hydroxyphenyl acetate, homovanillic acid (3-methoxy-4-hydroxyphenylacetic acid), and recently most widely used Amplex Red (10-acetyl-3,7-dihydrophenoxazine) [39, 42]. These compounds reduce hydrogen peroxide to water, and the oxidized products are detected. Again, the assays are complicated by direct interaction of dyes with the enzyme redox components, and many controls are needed for reliable quantitative determination of hydrogen peroxide.
MITOCHONDRIAL ROS-PRODUCING ENZYMES
Mitochondria isolated from various tissues produce hydrogen peroxide upon aerobic oxidation of substrates [7, 41, 43]. The observed rates of the mitochondrial hydrogen peroxide production depend on the tissue, ionic composition and pH of the incubation media, and on the methods used for detection of ROS [44-46]. It strongly depends on the metabolic state: the production of hydrogen peroxide is maximal at state 4 oxidation of succinate, and it is greatly decreased in the presence of uncoupler or ADP (state 3) [7, 9, 47, 48]. According to Boveris et al. [9], the total rate of the H2O2 production was calculated to be on the order of 90 nmol/min per g wet weight of liver, and about 15% of that was attributed to mitochondria. Unfortunately, the paper by Boveris et al. [9] published many years ago (in 1972) is the only one where an attempt to approximate quantitatively the relative contribution of different cellular components to the total hydrogen peroxide production was undertaken. Higher relative contribution of mitochondria to the total ROS production in heart is expected because of their higher content as compared to liver. Up to 3% of the total oxygen consumed by mitochondria is converted to hydrogen peroxide [44, 48]. Respiratory chain-linked ROS generation was originally reported by P. Jensen (see Table 1), who observed antimycin A-insensitive1 oxygen consumption by mitochondrial membranes oxidizing NADH or succinate coupled with hydrogen peroxide production [4]. Further studies have identified the specific components of the respiratory chain responsible for ROS production – complex I [49-52] and complex III [16, 53, 54], and also a number of other mitochondrial oxidoreductases producing hydrogen peroxide and/or superoxide radical [55].
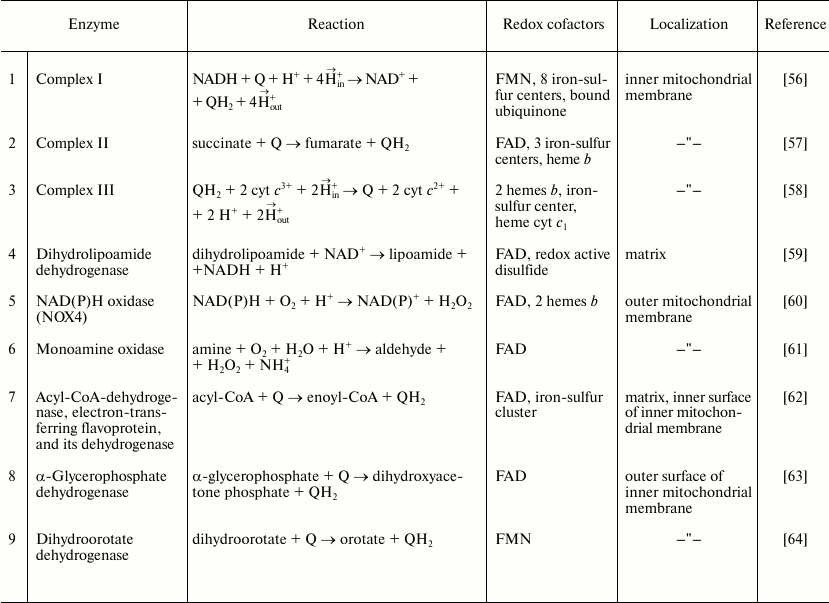
1 Antimycin A is a specific electron transfer inhibitor between ubiquinol and cytochrome c. The mitochondrial enzymes capable of ROS production are listed in Table 2. Their content and specific activities in different tissues vary greatly. The enzymes 1-4 and 7 are highly active in mitochondria from all tissues studied so far, whereas the specific activity of α-glycerophosphate dehydrogenase (8) is particularly high in brown adipose tissue and insect mitochondria. Limited or controversial information is available about the specific activities of enzymes 5, 6, and 9. Here we will describe some properties of enzymes 1-4 and 7, which are particularly active in heart mitochondria, and only short notes about some properties of enzymes 5, 6, 8, and 9.
Table 2. Mitochondrial ROS producing
enzymes
Dihydroorotate dehydrogenase. Dihydroorotate dehydrogenase (DODH, dihydroorotate:ubiquinone oxidoreductase, EC 1.3.99.11) is a flavoprotein catalyzing a unique redox reaction in the de novo pyrimidine synthesis pathway. Mammalian DODH is located in the inner mitochondrial membrane (class II dihydroorotate dehydrogenases) [65-69]. The FMN-containing active site of the enzyme formed by a hydrophilic part of the protein is exposed to the intermembrane space, whereas a smaller domain bound to the membrane surface contains a hydrophobic channel that makes FMN accessible for ubiquinone [64, 70]. The N-terminal sequence of the protein is a functional unit required for its binding to the membrane surface and for interaction with ubiquinone [71]. DODH has been found in many organs, and its specific activity is particularly high in rapidly proliferating tissues (tumor cells, mucosal cells of the ileum and colon crypts, kidney cortex) [66, 68, 69]. In heart, DODH activity is about 1/15th of that of succinate dehydrogenase, the most active respiratory enzyme in heart and liver mitochondria [69]. The formation of superoxide during aerobic oxidation of dihydroorotate in the presence of cyanide was originally demonstrated in liver mitochondria [72], the observation later being confirmed for brain mitochondria and for the solubilized purified liver mitochondrial enzyme [73]. FMN was postulated as the source of superoxide. However, histochemical studies of mitochondria in heart and kidney cortex showed that during dihydroorotate oxidation, hydrogen peroxide (the product of superoxide dismutation) was accumulated at the matrix-exposed area of the inner membrane, and not in the intermembrane space as expected if flavin is the site of superoxide generation. Specific staining was decreased in the presence of brequinar, a specific enzyme inhibitor [68]. It should be noted that cyanide used in those experiments for inhibition of respiratory activity significantly increases the level of DODH reduction, as well as that of other respiratory chain components capable of superoxide and/or hydrogen peroxide formation, so the amount of DODH-specific ROS production could be overestimated. Whether DODH effectively produces ROS in the absence of the respiratory inhibitors is not yet clear [70]. It has been shown recently that some drugs specifically interacting with the hydrophobic channel of the enzyme, thus preventing FMN-ubiquinone oxidoreduction, significantly decrease ROS formation in cancer cell cultures [74].
Mitochondrial glycerophosphate dehydrogenase. The mitochondrial α-glycerophosphate dehydrogenase (mGPDH, type 2 GPDH, α-glycerol-3-phosphate:ubiquinone oxidoreductase, EC 1.1.99.5) is located on the outer surface of the inner mitochondrial membrane [75]. It is a FAD-containing protein catalyzing oxidation of cytoplasmic α-glycerophosphate by ubiquinone. When operating together with cytoplasmic enzyme (NAD+-dependent GPDH, type 1), mGPDH catalyzes the transfer of reducing equivalents from cytosolic NADH to the mitochondrial respiratory chain (glycerophosphate shuttle). The mitochondrial enzyme is widely distributed, although its activity and content in different tissue vary greatly. Brown adipose tissue, placenta, pancreatic islets of Langerhans, testis, type II skeletal muscle, and brain are tissues with high mGPDH activity, whereas heart, liver, and kidney mitochondria show low activity [76, 77]. The ratio of mGPDH activity to that of succinate dehydrogenase in heart mitochondria is only 1/15 [76]. The enzyme content in the tissues with its low activity is strongly increased by thyroid hormones [78]. The atomic structure of the mitochondrial enzyme has not been established, but comparative analysis of cDNA coding bacterial, yeast, and higher eukaryote enzymes reveals high homology of the protein amino acid sequences [79]. The three-dimensional structure of E. coli type 2 GPDH [80] appears as a dimer in which two bound monomers form a “cap” protruding to the bacterial cytoplasm. The “cap” isolates the substrate dehydrogenating active FAD-containing sites from the aqueous environment. Each monomer has two domains, a hydrophilic participating in the “cap” formation (C-end) and an N-terminal FAD-binding domain. A positively charged area of the protein located at the membrane surface binds to negatively charged plasma membrane phospholipids. No sequences characteristic for transmembrane helices are found in the primary structure, thus suggesting that the enzyme is bound to the membrane surface [80]. An EF-motif is found in the primary structure [79] – an observation that is in accord with known stimulatory effect of Ca2+ on the enzyme activity (decrease in Km for α-glycerophosphate) [75]. Heart mitochondria generated ROS during coupled (state 4) α-glycerophosphate oxidation at the rate of only 3 pmol/min per mg of protein, whereas the activity seen under the same conditions with succinate as the substrate was about 260 pmol/min per mg of protein [76]. The glycerophosphate-supported ROS production is increased by the inhibitors of complex III, antimycin A or myxothiazol. In the presence of antimycin A, hydrogen peroxide is produced at several sites when α-glycerophosphate is oxidized: those are GPDH itself and also complexes II and III, which are reduced by QH2 [76, 77]. Only 6% of the total generation was originated from mGPDH under these conditions [77]. What particular redox components of the enzyme react with oxygen is not clear. Reduced ubiquinone or protein-bound ubisemiquinone have been proposed as the source [40, 77]. Strong stimulation of mGPDH-mediated ROS production by the one-electron acceptor ferricyanide [81] was observed.
NAD(P)H oxidase (NOX4). NAD(P)H oxidases of the NOX family (EC 1.6.3-) are enzymes that generate ROS as the product of their natural catalytic cycle. They are the membrane-bound proteins catalyzing electron transfer from NAD(P)H to oxygen, thus producing superoxide. NAD(P)H oxidase of phagocytes (NOX2) responsible for so-called “respiratory burst” is the best-studied member of the family [60]. No atomic structure of any member of the family is available. The sequences, hydropathy profiles, and immunological analysis suggest that all NAD(P)H oxidases are structurally similar. They are integral membrane proteins with six transmembrane helices and five hydrophilic loops: three on one side of the membrane and two on the other side [60]. The NAD(P)H- and FAD-binding sites are located in the C-terminal region. Heme b is bound to the transmembrane helices. The sequential intramolecular transmembrane electron transfer NAD(P)H → FAD → heme b → heme b → O2 results in a formation of two molecules of superoxide, which are the true, not side product of the enzyme. Mitochondria contain isoform NOX4, which requires only the one additional membrane protein p22 for activation, whereas other NOXs need, in addition, several cytoplasmic proteins [60, 82]. In contrast to other members of the family, the product of NOX4 is hydrogen peroxide, not superoxide, although oxygen is reduced by a one-electron donor (heme b). A histidine residue located in the E-loop serves as the donor of protons required for rapid catalytic dismutation of the product [83]. NOX4 was first found in kidney (originally named Renox [84]), and later the enzyme was detected in the mitochondrial fraction of cardiomyocytes [85, 86]. The intramitochondrial location and topology of NOX4 have not been defined [87].
The physiological function of NOX4 in heart is unclear. An increase in its expression during aging and heart hypertrophy has been reported [85]. An increase in NOX4 activity positively correlates with increased oxidation of SH-groups in several subunits of complex I and inactivation of aconitase and citrate synthase leading to a disorder of the mitochondria and cell death [85, 86].
Monoamine oxidase. Monoamine oxidase (MAO, EC 1.4.3.4) is a FAD-containing enzyme that controls the level of the neurotransmitters and protects cells from deteriorating effects of amines. MAO is located in the outer mitochondrial membrane as a dimer formed by hydrophobic transmembrane helices [61, 88-90]. Two homologous isoforms of MAO exist (MAO-A and MAO-B) that differ in their specificity and sensitivity to inhibitors. MAO-A preferentially oxidizes norepinephrine and serotonin, whereas phenylethylamine and benzylamine are the best substrates for MAO-B. Either isoform deaminates dopamine, tyramine, epinephrine, and tryptamine at the same rates [61, 89, 90]. Hydrogen peroxide is the product. The only redox component, FAD, is covalently attached to a cysteine residue of the polypeptide chain [91]. The enzyme is found in all tissues (nervous tissue, heart, lungs, liver, blood vessels, etc.) [92]. In cardiomyocytes, MAO-A is the major specie [92]. The reaction products, hydrogen peroxide, aldehydes, and ammonium, are toxic. Increased expression of the enzymes or their activation result in a number of pathologies. According to several reports, MAO plays the major role in the development of oxidative stress of the nervous system and heart. Particularly, deteriorating effect of reperfusion after ischemia was reported to be caused by MAO-A [93]. Serotonin is accumulated during an ischemic period, and when oxygen becomes available upon reperfusion MAO produces hydrogen peroxide. The latter gives rise to peroxidation of mitochondrial phospholipids and apoptosis. Inhibitors of MAO are cardioprotective. They decreased hydrogen peroxide production and prevented reperfusion-induced destructions, such as myofibril hypercontraction, interstitial edema, and swelling of mitochondria [94]. The addition of serotonin to cardiomyocyte culture [94] resulted in a decrease in reduced glutathione content and in increase in hydrogen peroxide production. Also, increased expression of proapoptotic (BAX) protein, decrease in antiapoptotic (Bcl-2) protein, and release of cytochrome c to the cytoplasm were observed. The inhibitors of MAO-A prevented these serotonin-induced effects [94]. Other observations that suggest an important role of MAO in development of myocardial oxidative stress is its increased expression during aging, which correlates with an inhibitor-sensitive increase in hydrogen peroxide [95].
Acyl-CoA dehydrogenase, electron-transferring flavoprotein, and its dehydrogenase. The electron-transferring flavoprotein (ETF) discovered by Crane and Beinert [96] serves as the only electron acceptor for nine different mitochondrial FAD-containing acyl-CoA dehydrogenases [62, 97]. This heteromeric protein contains one molecule of FAD per dimer located in a cleft between the α- and β-subunits [98]. Two ETF molecules are apparently needed for oxidation of the reduced acyl-CoA dehydrogenases because FAD in the catalytic cycle acts as a one-electron acceptor [99]. The reduced ETF transfer electrons to ETF:ubiquinone oxidoreductase (EC 1.5.5.1) [100], an enzyme which functionally is a member of the mitochondrial respiratory chain. This is a single-subunit protein located on the inner side of the inner mitochondrial membrane. One FAD and a [4Fe-4S] iron-sulfur cluster, which act as one-electron acceptors/donors, have been detected in the enzyme [62, 101]. ROS production during palmitoyl carnitine (penetrating form of fatty acids) oxidation in mitochondria from skeletal muscles and brown adipose tissue has been observed [46, 102, 103]. What the particular component that generates ROS during fatty acid oxidation – ETF, ETF:ubiquinone oxidoreductase, complex III, or complex I – is not clear [46, 102, 104]. Participation of acyl-CoA dehydrogenases themselves [103] in ROS generation cannot be excluded because their reduced substrate-free forms rapidly reduce oxygen to hydrogen peroxide [105].
Respiratory complex I. The generation of ROS by complex I will be described in more details than for other enzymes because it is the major component of the respiratory chain-mediated reaction. The mitochondrial proton-translocating NADH:ubiquinone oxidoreductase (complex I, first coupling site, EC 1.6.99.3) and its functional homolog, prokaryotic type 1 NADH dehydrogenase, catalyze oxidation of NADH by quinones coupled with translocation of four protons per mole of NADH oxidized, thus building up Δp across the membrane. The reaction is reversible, and NAD+ reduction is observed if ubiquinone is reduced by any other quinone-reducing dehydrogenase when the membrane is energized (reverse electron transfer). The Δp-dependent reduction of the enzyme redox components in the absence of NAD+ under aerobic conditions results in ROS generation.
The structure of the enzyme is extremely complex. The mammalian complex I is composed of 44 individual subunits (total molecular mass about 980 kDa) [106, 107], in Yarrowia lipolytica mitochondria 42 different subunits (molecular mass ~947 kDa) have been identified [108, 109], whereas prokaryotic type 1 dehydrogenase contains 13-15 subunits (molecular mass ~550 kDa) highly homologous to the corresponding subunits of the eukaryotic enzymes. The type 1 prokaryotic enzyme represents the minimal structural requirement for the overall proton-translocating NADH:ubiquinone reductase reaction [110, 111]. The enzymes from different species have the same set of redox components. They are FMN [112], 8-9 iron-sulfur clusters [113-116], and tightly bound ubiquinone [117]. The functional roles of the so-called “supernumerary” subunits of the eukaryotic complexes are unknown.
Complexes I from various sources and prokaryotic homologs are similar in their L-shape arrangement (Fig. 3a) [111, 118-120]. The peripheral (almost vertical) part (about 150 Å) protrudes into the mitochondrial matrix (or bacterial cytoplasm). It is composed of relatively hydrophilic subunits. The FMN (primary electron acceptor) and NADH-binding site are located far from the membrane plane and exposed to the aqueous environment. All the redox components of the Thermus thermophilus [120-122] and eukaryotic Yarrowia lipolytica enzymes [123] were visualized in subunits of the protruding hydrophilic part (Fig. 3a). The iron-sulfur centers (designated by the letter N and numbered according to Ohnishi’s nomenclature [124]) form an electron-conducting “wire” connecting the flavin and the ubiquinone-binding site, which is located at 25-30 Å distance from the membrane [119]. They transfer electrons in the sequence: NADH → FMN → N3 → N1b → N4 → N5 → N6a → N6b → N2 → Q [118, 120]. Center N1a is located out of the “wire” in close (about 13 Å) vicinity of flavin (Fig. 3a). NADH is an obligatory two-electron donor, and FMN and N1a apparently operate as the “coupling” pair to provide rapid oxidoreduction between two-electron donor (NADH) and one-electron acceptor (N3), as suggested for a number of other iron-sulfur containing flavoproteins. Three transmembrane subunits homologous to bacterial Na+/H+ antiporters located in the horizontal part of the structure shown in Fig. 3a have been recently identified in the structure [125]. They are believed to catalyze redox-driven proton translocation. The large distance between all redox components and putative proton translocating subunits is suggestive of a redox-dependent conformational pump mechanism operating at coupling site 1 [126].
Fig. 3. Mitochondrial complex I. a) Intramolecular electron transfer (midpoint potentials of the substrates (NADH and ubiquinone) and the iron-sulfur clusters [118] are indicated in brackets). Possible sites of ROS generation are marked by asterisks. b, c) Relative contribution of superoxide (open bars) and hydrogen peroxide (filled bars) to the overall ROS production by SMP-bound (b) and purified (c) complex I [127].
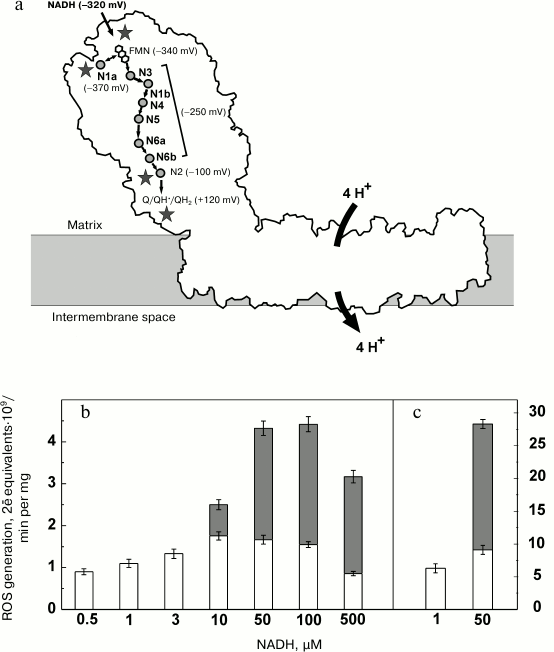
Bovine heart SMP generate ROS at the rate of 2-2.5 (2ē equivalents⋅109/min per mg of protein)2 when they catalyze controlled (state 4) oxidation of succinate or NADH, the specific activity corresponding to about 0.25% of the total oxidase [127]. During the NADH oxidase reaction, about 85% of ROS is produced by complex I, and the residual 15% is generated by respiratory complexes II and III [127]. Complex I operating within tightly coupled SMP is capable of ROS production in several assay systems: 1) energy-linked reverse electron transfer (in the absence of NAD+); 2) the standard overall NADH oxidase; and 3) oxidation of NADH in the presence of respiratory inhibitors (rotenone, piericidin, antimycin A, etc.) [7, 9, 49, 51, 52, 127-130]. It has been reported that complex I produces only superoxide, the precursor of hydrogen peroxide [55]. This observation is in accord with a recent report by Hirst’s group [131], who has shown that during oxidation of very low NADH concentrations by purified bovine heart complex I 90% of the electron flow produces superoxide and only 10% results in hydrogen peroxide formation [131]. Purified E. coli enzyme produced 80% of ROS as hydrogen peroxide [132]. Recently, we have shown that the membrane-bound complex I (SMP) as well as purified enzyme is capable hydrogen peroxide and superoxide production (Fig. 3b) [127, 133]. The rate and partitioning between the products apparently depend on the enzyme preparations used (intact mitochondria, SMP, isolated enzyme) and on the assay conditions (substrate concentration, ionic composition, and pH).
2 2ē equivalents are equal to 1 mol of NADH oxidized. This way to express ROS producing activities is used because the formation of superoxide or hydrogen peroxide requires one or two electrons, respectively. The reaction catalyzed by intact mitochondria or coupled SMP when it proceeds via reverse electron transfer is strongly dependent on the membrane energization; it is inhibited by ADP (state 3 respiration) and very sensitive to uncoupling [7, 9, 49, 51, 127-129]. The respiratory control ratio correlates with the rate of aerobic succinate-supported reverse electron transfer [134]. The better coupled SMP are, the higher the rate of ROS generation by complex I and the higher the fraction of hydrogen peroxide in the total ROS produced is (35 and 60% at respiratory control ratio of 4.5 and 8.0, respectively) [127]. The rate of ROS production and fractional products formation during NADH oxidation are less sensitive to the membrane energization [127-129]. The respiratory inhibitors increase superoxide and hydrogen peroxide production up to 2-fold, thus indicating that the level of reduction of the respiratory chain components rather than the membrane energization is the key parameter that determines ROS production [50, 51, 127, 129].
The partitioning between superoxide and hydrogen peroxide by complex I also depends on NADH concentration. The rate of superoxide production reaches a maximum at 10-50 µM NADH and gradually decreased in the millimolar range of the substrate concentration [127, 129]. The apparent KmNADH as determined from a linear double-reciprocal plot for the ascending part of the superoxide production titration curve is as low as about 0.5 µM [127]. The apparent Km value for the substrate-donor in the reaction catalyzed by any multi-redox-component oxidoreductase sequentially transferring electrons from the donor to an acceptor depends on the redox potential difference between the primary electron acceptor (FMN for complex I) and the component that reacts with an acceptor. The larger the difference is, the lower the apparent Km is observed [135]. Very low KmNADH for superoxide production [127, 131] indicates that a component immediately reacting with oxygen has significantly higher redox potential than that of FMN. The iron-sulfur cluster N2, the most positive redox component of complex I with midpoint potential of about –100 mV seems likely to react with oxygen. This cluster is located in close vicinity to the ubiquinone-binding site at the funnel-like area at the distance of 25-30 Å from the membrane plane [119]. All other iron-sulfur centers are well insulated by the protein. Remarkably, all the specific inhibitors of complex I act at either the entry point (competitive inhibitors such as NADH-OH [136, 137] and ADP-ribose [138] or diphenyleneiodonium [139, 140]) or at the exit site (many inhibitors of the Q reduction site [141-143]).
Hydrogen peroxide formation depends on NADH concentration quite differently. At low substrate concentrations (up to 3 µM) no production is seen, and the rate reaches a constant value at higher (up to millimolar) range of NADH concentration where its relative contribution to the overall ROS production is about 60%. The half-maximal rate is observed at much higher (50-fold) NADH concentration than that for superoxide production (~25 µM; Fig. 3b) [127].
Complex I-catalyzed ROS production is inhibited by µmolar NAD+ concentrations [129]. The specific activity of complex I in ROS generation is less than 1% of its major oxidase or NADH:artificial acceptors reductase reactions, so it is safe to assume that all redox components are in equilibrium with the NAD+/NADH couple during the steady-state reaction (the kinetic term contribution to apparent Km is negligible). Thus the dependence of ROS production on NAD+/NADH ratio can be used to approximate the midpoint redox potential of the component reacting with oxygen. The NAD+/NADH ratios reported in the literature for half-maximal ROS production by complex I are greatly variable (from 0.01 up to 7.0 [52, 131, 144-146], see Table 2 in [146]). At least two possible reasons for these variations are conceivable. First, in intact mitochondria other than complex I enzymes that are also in equilibrium with the NAD+/NADH couple produce ROS, for example, the E3 component of α-oxoglutarate and pyruvate dehydrogenases [35]. Second, the titration of complex I as well as other enzymes by the NAD+/NADH couple is not a true redox titration because the relative binding affinities of the reduced and oxidized enzyme to NAD+ and NADH may significantly contribute to the apparent NAD+/NADH ratio required for ROS production. Thus (NAD+/NADH)0.5 value depends on the total nucleotide pool concentration, and it decreases from 0.2 to 0.05 when the total NAD+ plus NADH concentration increases from 50 to 500 µM [146]. Lower value of (NAD+/NADH)0.5 is expected for intact mitochondria because the total concentration of pyridine nucleotides in the mitochondrial matrix is much higher (~4-7 mM). Indeed, Kushnareva et al. reported (NAD+/NADH)0.5 ratio of 0.01 for heart mitochondria as deduced from titration by the acetoacetate/β-hydroxybutyrate couple [144]. They hypothesized that iron-sulfur cluster N1a is the center of superoxide formation, since half-maximal production in their experiments was seen at –392 mV, a value close to the midpoint potential of N1a (–370 mV [111, 150]). Their interpretation is in accord with Sazanov’s proposal on participation of N1a and FMN/FMN• in the catalytic cycle [118].
It has been shown in earlier studies that the rate of superoxide production by SMP is less dependent on the medium redox potential than on the nature of the substrate-nucleotides. Half-maximal rate was detected at approximately the same ratio of NAD+/NADH and acetyl-NAD+/acetyl-NADH, whereas the redox potentials of these couples differ by about 60 mV [52].
Hydrogen peroxide and superoxide production by complex I depend on NAD+/NADH differently. At low nucleotides concentration (50 µM), which is optimal for either species, the rate of H2O2 production satisfactorily fits the Nernst equation for a two-electron reaction with midpoint redox potential of –350 mV ((NAD+/NADH)0.5 = 0.13) [127]. This value is close to the midpoint potential of FMN (–370 mV at pH 8.0) estimated from EPR data [147]. The same dependence for superoxide production does not fit the Nernst equation, and half-maximal activity is observed at significantly higher NAD+/NADH ratio (0.33) [127]. Superoxide production does proceed even when the pool of nucleotides is 90% oxidized, i.e. at a potential that is much more positive than that of the FMN/FMNH– couple. These results also indicate that the most positive N2 center is the source of superoxide production.
The dependence of superoxide production by purified complex I on NAD+/NADH ratio was also measured by Kussmaul and Hirst, who observed maximal activity at potential less than –400 mV, whereas no generation was seen at potential higher than –300 mV [131]. Their titration curve fitted the theoretical curve of two-electron titration of the FMNH–/FMN couple with midpoint potential of –360 mV.
Galkin and Brandt [151] used an apparently direct approach that excluded iron-sulfur cluster N2 as the site of one-electron reduction of oxygen. They found that a mutant form of Y. lipolytica isolated complex I (R141M), which showed no EPR-detectable center N2, produced superoxide with the same rate (96%) as did the enzyme from the wild strain. The data on superoxide formation activity catalyzed by either form were normalized to their NADH:artificial acceptor reductase activities. However, the same normalization of their data to the “natural” activities shows that the “mutant form” was 60% active in quinone reductase activity (see Table III in their paper [151]). The authors did not report the data on rotenone-sensitivity of the quinone reductase activities. If they were rotenone-sensitive, the data would suggest that the R141M mutant catalyzed “normal” quinone reductase reaction without participation of N2, a possibility that seems unlikely. We believe that exclusion of iron-sulfur cluster N2 as a possible site of superoxide production should wait for more experimental verification.
Brand et al. measured complex I-mediated ROS production during forward and reverse electron transfer in intact skeletal muscle mitochondria [152]. They concluded that there are two sites of generation – flavin and bound ubisemiquinone. In their view, in the forward reaction flavin equilibrated by the NAD+/NADH couple is the site of ROS, whereas bound ubisemiquinone equilibrated by the Q/QH2 couple produces superoxide during the reverse reaction. This proposal is similar to that of Ohnishi et al. [153]. They propose that ROS are generated by either flavin or by ubisemiquinone, which are in dynamic equilibrium dependent on the membrane energization and the degree of ubiquinone reduction [153].
ROS production by complex I in intact mitochondria with succinate as the substrate is substantially higher (3-4-fold) than that observed with NAD+-dependent substrates. This would seem trivial, because the reaction is inhibited by high NADH level, and the reduction of the pyridine nucleotide pool by NAD+-dependent substrates is expected to be higher than that in the presence of succinate. This interpretation, however, does not seem valid: the degree of NAD+ reduction in tightly coupled mitochondria from various tissues oxidizing succinate is always higher than that observed upon oxidation of NAD+-dependent substrates. This puzzling phenomenon originally observed by Chance and Hollunger as early as in 1961 [154] is still not satisfactorily explained. They proposed “compartmentalization” of the intramitochondrial pyridine nucleotides as a possibility [154], but this has no direct experimental evidence. In numerous studies on mitochondrial ROS production, succinate is most frequently used as the respiratory substrate. It should be emphasized that coupled mitochondria oxidizing externally added succinate cannot be considered as the model of any physiologically conceivable state. Succinate, an intermediate of the Krebs cycle, is produced and utilized in the mitochondrial matrix, providing one fifth of the reducing equivalents during complete oxidation of pyruvate. No other quantitatively significant cytoplasmic sources of succinate exist in aerobic metabolic pathways (here we put aside α-oxoglutarate-dependent proline hydroxylation [155, 156] and succinic semialdehyde transformation [157] as insignificant for the total respiratory activity of mammalian tissues). This does not mean that the reverse electron transfer catalyzed by complex I does not contribute to reduction of the intramitochondrial pyridine nucleotides. Ubiquinol, the actual substrate for the energy-linked reverse electron transfer, is produced in several mitochondrial metabolic pathways, such as fatty acid β-oxidation or oxidation of α-glycerophosphate.
The well-known “burst” of ROS formation upon anaerobic–aerobic state transition, such as organ reperfusion after ischemia, is a phenomenon that seems relevant to the present discussion. The mechanistic reason(s) for the burst is(are) not clear. Complex I may contribute to “burst”. If either NADH or oxidized ubiquinone is not available, the transformation of the enzyme to so-called de-active state is observed (at T > 30°C), and electron transfer from N2 to ubiquinone becomes blocked (see papers [38, 158, 159] and reviews [160, 161]). The active (A) state to de-active (D) state transition has been detected for isolated complex I [158, 159], SMP [38], intact heart mitochondria [36], and in ex vivo studies of perfused hearts [162]. De-activation of complex I is equivalent to the inhibition by rotenone, which is known to increase ROS production. The back transformation of D-form to A-form is a slow process, which is inhibited by free fatty acids and divalent metals cations [163-165]. Taken together, the data on A/D-transformation suggests the following scenario of the normal state → ischemia → reperfusion transition. A certain rate of complex I-mediated ROS production under the initial normal state occurs. It stops under ischemia because no oxygen is present. A sudden increase in oxygen up to the normal level would result in a burst of ROS because de-activated enzyme will be directly oxidized by oxygen, not by ubiquinone. An increased ROS production is expected for the time needed for slow the D-to-A transformation and restoration of the normal ubiquinone reductase activity. De-activation of complex I is accompanied by exposure of a hidden SH-group (Cys39) located in the ND3 subunit [166], and if this cysteine residue is covalently modified the D-to-A transition is prevented [167, 168]. Reversible modification of the specific SH-group may be explored for possible pharmacological protection against reperfusion-induced burst of ROS [169]. Such a protective effect of a mitochondrially-targeted nitrosylating compound has recently been reported [170].
Complex II. Complex II (succinate dehydrogenase, succinate:ubiquinone oxidoreductase, EC 1.3.99.1) is a marker enzyme of the inner mitochondrial membrane. It catalyzes one of the Krebs cycle reactions and supplies electrons to the respiratory chain. Atomic structures of complex II and its homolog, fumarate reductase, have been determined [57, 171]. The enzymes are composed of four subunits. The hydrophilic part exposed to the mitochondrial matrix or bacterial cytoplasm is formed by FAD-containing subunit SDHA. The SDHB subunit bears three iron-sulfur clusters. The SDHC and SDHD subunits are hydrophobic and immersed into the membrane. They contain heme b and the ubiquinone-binding site [57, 171]. Superoxide production by purified bovine heart succinate dehydrogenase and also by isolated and reconstituted complex II has been reported [172]. The rates of superoxide and hydrogen peroxide formation by E. coli succinate dehydrogenase and fumarate reductase catalyzing the reduction of fumarate by menaquinol were measured [173]. The dependence of succinate-supported ROS formation by fumarate reductase on succinate concentration is bell-shaped [173]. At low substrate concentration the enzyme produced mostly superoxide, whereas at higher succinate concentration hydrogen peroxide was mostly produced. These results have been interpreted to suggest that superoxide is produced by flavin when the second electron from the FAD radical can be donated to the adjacent [2Fe-2S] iron-sulfur cluster. If the latter is reduced (at higher succinate concentration), the second electron is donated to preformed superoxide, and hydrogen peroxide is thus produced. Escherichia coli succinate dehydrogenase, in contrast to fumarate reductase, generates only superoxide at 25-fold lower rate. The difference between the products and rates of the ROS production mediated by the enzymes apparently lies in the redox potentials of their cofactors [171]. Iron-sulfur cluster [3Fe-4S] in succinate dehydrogenase has significantly higher redox potentials than in its counterpart. In contrast, the redox potential of FAD and closely located [2Fe-2S] center of fumarate reductase are higher than corresponding values for E. coli succinate dehydrogenase. Under the steady-state, the mostly reduced components of succinate dehydrogenase are [3Fe-4S] cluster and heme b, whereas FAD and [2Fe-2S] are the mostly reduced components in fumarate reductase [171].
The skeletal muscle mitochondrial complex II has recently been reported to produce superoxide and/or hydrogen peroxide at relatively high rates [174]. The specific rate versus succinate concentration dependence was also bell-shaped with a maximum at presumably physiological (~0.4 mM) substrate concentration. Complex II also produces ROS if ubiquinone is reduced by mGPDH [174]. When the reaction was supported by mGPDH, complex II-supported ROS generation was sensitive to malonate, a competitive inhibitor of succinate binding, and atpenin, a Q-binding-site inhibitor [174]. Recently published data indicate that complex II is partially responsible for the succinate-supported ROS generation in the presence of rotenone [175] – activity previously ascribed as complex III-dependent.
Complex III. The mitochondrial ubiquinol:cytochrome c oxidoreductase (complex III, b-c1 complex, coupling site 2, EC 1.10.2.2) catalyzes oxidation of ubiquinol by cytochrome c coupled with transmembrane proton translocation and Δp formation. In prokaryotes, the reaction is catalyzed by a three-subunit “minimal” complex composed of cytochrome b, cytochrome c1, and iron-sulfur protein [176, 177]. The mitochondrial enzyme contains an additional 8 subunits [178-181]. The membrane-bound complex exists as a dimer reacting with cytochrome c [178-181]. The core of each monomer is formed by hydrophobic polypeptide bearing two hemes, low potential (bL) and high potential (bH) located closed to the outer and inner surface of the coupling membrane, respectively. Cytochrome c1 and iron-sulfur protein (Rieske protein) are hold by transmembrane helixes, and their peripheral domains where redox groups are located are exposed to hydrophilic intermembrane space. Rieske proteins provide cross interaction within the dimer; its membrane-located part is bound to one monomer, whereas the peripheral with the other one [58]. The protein is conformationally mobile; it occupies at least three distinct positions where its iron-sulfur cluster is located either in close vicinity to heme c1 (c1-position) or to heme bL (b-position) or in between [179, 180, 182].
The mechanism of complex III-catalyzed Δp formation (proton-motive Q cycle) was originally proposed by Mitchell [183, 184]. Its contemporary version is schematically depicted in Fig. 4. The first step is oxidation of ubiquinol at site Qo where the two electrons bifurcate, one reducing the iron-sulfur cluster of Rieske protein (reaction 1) and the other being transferred to heme bL (reaction 2). Two protons are released in the intermembrane space per ubiquinol oxidized at site Qo. The iron-sulfur cluster sequentially reduces cytochrome c1 and cytochrome c, the terminal electron acceptors (reactions 3 and 4). Reduced heme bL donates an electron to heme bH located at the opposite side of the membrane (electrogenic step of the overall reaction, reaction 5) in close vicinity to the other ubiquinone-binding site Qi. One-electron oxidation of heme bH by ubiquinone results in ubisemiquinone formation at Qi (reaction 6a). Two possible mechanisms of electron bifurcation at site Qo are under discussion [185]. The reactions 1 and 2 may proceed sequentially, thus producing an ubisemiquinone intermediate [186]. The alternative possibility is that cooperative, synchronous oxidation of ubiquinol by the iron-sulfur cluster and heme bL takes place without semiquinone formation [187-191]. In the second half-reaction, ubiquinol is oxidized at the Qo site followed by reactions 1-5, and ubisemiquinone is reduced at site Qi, coupled to uptake of two protons from the matrix space (reaction 6b). Oxidation of one ubiquinol molecule by two cytochrome c molecules and translocation two protons from the matrix to intermembrane space is the net result of the catalytic cycle.
Fig. 4. Proton motive force generation by complex III. See text for explanation.
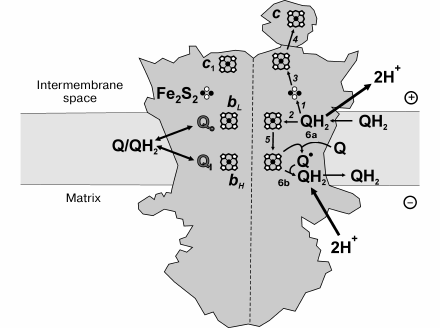
The ubiquinol:cytochrome c reductase reaction is reversible [189], and special mechanisms exist to prevent some undesirable short circuiting reactions [190]. The obligatory bifurcation of electron flow at site Qo required for the proton translocation is achieved by the movement of the iron-sulfur cluster from b-to-c1 position [185, 192]. Another possibility is that ubiquinol at site Qo can be oxidized only when both iron-sulfur cluster and heme bL are oxidized, and ubiquinone reduction at site Qi proceeds only when these redox components are reduced [189, 190]. The dimeric structure that operates by the half-of-the-sites reactivity mechanism apparently plays an important role in prevention of electron short circuiting reactions [193-195].
The site of ROS production by complex III is believed to be ubisemiquinone formed at site Qo [185, 196, 197]. The rate of superoxide production by complex III is substantially lower than that mediated by complex I. The ROS production by purified complex III is increased by heat treatment or by protein kinase K digestion [198]. Knock-out of the cytochrome c1 encoding gene increases ROS production, whereas mutations that restrict conformational mobility of Rieske protein result in decreased activity [199, 200]. Antimycin A and other Qi-site inhibitors strongly stimulate superoxide generation [53]. Also, stimulation is seen if electrogenic electron transfer between hemes bL and bH across the coupling membrane is limited by Δp [201]. The rate of superoxide formation is maximal if 25-30% of the mitochondrial ubiquinone is oxidized [202]. It has been proposed that superoxide is formed when ubiquinone binds at site Qo and is reduced to ubisemiquinone by heme bL in the reverse electron transfer reaction [185, 196, 202]. This proposal has been experimentally confirmed in studies of mutants of Rhodobacter capsulatus [199, 200].
Another mechanism of superoxide production by complex III has been proposed by Yu et al. [198, 203]. They have shown that the ubiquinol:cytochrome c reductase activity of R. sphaeroides complex III is decreased about 2-fold due to a decreased rate of heme bL reduction when the enzyme operates under anaerobic conditions [203]. According to the authors, oxygen participates in the catalytic activity by accepting an electron from ubiquinol, and protonated superoxide donates an electron to heme bL. This proposal agrees with data showing the presence of a cavity located between heme bL and the ubiquinol-binding site that binds a xenon molecule, imitating oxygen binding [203].
Complex III generates superoxide at both sides of the membrane [46, 204], this observation apparently contradicting the hypothesis on superoxide formation at site Qo. To explain this phenomenon, Muller et al. [204] proposed that protonated superoxide generated at site Qo leaves the site via a hydrophobic channel, which provides equal distribution of the product between the inner and outer space.
Dihydrolipoamide dehydrogenase. Dihydrolipoamide dehydrogenase (DLDH, lipoamide dehydrogenase, lipoamide reductase, Straub diaphorase, E3, EC 1.8.1.4) is a member of large family of flavin-containing thiol-disulfide oxidoreductases including glutathione-, mercury, and thioredoxin reductases. Some of these proteins, such as glutathione reductase, are the most-studied redox enzymes and are used in biochemistry textbooks to illustrate several aspects of biological catalysis [205]. DLDH is ascribed as a moonlighting protein capable of, besides its major function, several non-canonical activities such as proteolytic [206], quinone reductase [207], acyltransferase [208], and recently rediscovered ammonium-dependent hydrogen peroxide generation [209]. The enzyme is an FAD-containing component of the complexes catalyzing the terminal step in oxidative decarboxylation of α-oxoglutarate, pyruvate, branched-chain oxoacids [210, 211], and glycine [212], i.e. NADH reduction by the dihydrolipoamide moiety covalently bound to the dihydrolipoamide acyl transferase protein (E2-component of the above-listed holoenzymes). Homologs of DLDH are widespread in bacteria, fungi, and higher eukaryotes [213]. Free DLDH was found in the mitochondrial matrix along with the bound form [214]. Surprisingly, DLDH has been found in some organisms lacking of NAD+-dependent dehydrogenases of α-oxoacids [215, 216].
The enzyme is a homodimer of 51-kDa subunits. The active site of each monomer is made up of a pyridine nucleotide-binding region, where FAD and catalytically active disulfide are located [213, 217]. The reduction of the active site disulfide bond by dihydrolipoamide, sequential FAD reduction, and hydride transfer from the reduced flavin to NAD+ are the catalytic steps of the overall reaction [218, 219]. These steps are readily reversible, in contrast to the irreversible decarboxylation step of the overall dehydrogenase reaction. Isolated DLDH catalyzes NAD+ reduction by dihydrolipoamide as well as NADH oxidation by oxidized lipoamide with comparable turnover numbers.
Intact or permeabilized brain mitochondria have been shown to catalyze α-oxoglutarate oxidation-supported ROS production [220]. Purified cardiac α-oxoglutarate and pyruvate dehydrogenases are capable of superoxide [221] and/or hydrogen peroxide production [220, 222, 223]. The significance of DLDH-mediated mitochondrial ROS production was supported by studies of heterozygous knock-out mice deficient in DLDH (DLDH+/–) [220]. Their mitochondria showed 2-fold lower ROS production during α-oxoglutarate oxidation [220]. Although the rates of oxoacid oxidation by purified α-oxoglutarate dehydrogenase complex (OGDHc) and pyruvate dehydrogenase complex (PDHc) in the presence of NAD+ are almost the same, the rate of ROS production in the presence of α-oxoglutarate is about twice that observed in the presence of pyruvate [220]. ROS production by purified OGDHc is reciprocally controlled by NAD+/NADH ratio: when it is low, the rate of oxoacid oxidation is low and H2O2 production is high [223].
About half of hydrogen peroxide produced by permeabilized heart mitochondria during NADH oxidation originates from complex I, and the other half is produced by DLDH [35]. DLDH-catalyzed production is strongly (more than 5-fold) stimulated by millimolar concentrations of ammonium, and up to 90% of the total production seen in the presence of ammonium originates from DLDH [35]. Purified bovine and pig heart DLDH and the recombinant human heart enzyme also catalyze NADH-supported ammonium-dependent hydrogen peroxide production, whereas their “physiological” NADH:lipoamide reductase activity is insensitive to ammonium [224]. No data on the significance of ammonium stimulation under physiologically relevant conditions are available. It is expected that operation of ammonium-producing intramitochondrial enzymes such as glutaminase, glutamate dehydrogenase, AMP deaminase would increase generation of ROS. Poorly understood toxicity of ammonium may at least be partially due to its activating effect on DLDH-supported ROS production.
The role of OGDHc in oxidative stress is under vivid discussion (see reviews [225-227]). Being the source of hydrogen peroxide, the enzyme itself is damaged by ROS [221].
NOTES ON PHYSIOLOGICAL AND PATHOPHYSIOLOGICAL SIGNIFICANCE OF
MITOCHONDRIAL ROS PRODUCTION
An increase in atmospheric oxygen in the history of the Earth has resulted in great improvement of energetics of life and, on the other hand, in the necessity for protection against undesired deteriorating oxidative reactions. Two strategies could be used to solve the problem. One is to develop individual protective mechanisms for many potentially reactive oxidoreductases containing flavins and/or iron-sulfur centers by the specific arrangement of their protein structure including additional subunits that have no other than protective functions to avoid any ROS production. The other possibility is to create a “general defense system” – specific enzymes rapidly converting inevitably formed ROS to water. It seems the latter alternative was used during evolution. Numerous experimental data show that almost all flavo- and iron-sulfur proteins, including those knowingly not operating as oxidases, do react with oxygen, thus producing either superoxide or hydrogen peroxide. In other words, their specific protection is not perfect. This is compensated by the widespread presence of SODs, peroxidases, and catalase even in mitochondria where ROS production under artificially “optimal” conditions is very small compare with their oxygen consuming activity. Does the mitochondrial ROS production have any physiological function? Numerous papers have recently appeared in the literature where signaling cascade reactions with the participation of hydrogen peroxide are suggested and discussed (see for example review [12]). Note should be made that signaling molecules are defined, in contrast to metabolites, as those that bind to a receptor and induce its structural change without chemical transformation of the signaling molecule. The structural change of a receptor then results in activation (or inhibition) of some metabolic pathways. Many hormones, second messengers (c-AMP, Ca2+) and protein factors are true signaling molecules. Certainly, if expanded (unjustified in our opinion), the meaning of “signaling molecule” term could be applied to any metabolite. The mechanism of hydrogen peroxide “signaling” postulated in most schemes is a trivial non-enzymatic oxidation of deprotonated sulfhydryl groups that results in their catalytic (or signaling) activities. An obvious shortcoming of those schemes is that no target protein-specific peroxidases are postulated. It appears that if a regulatory signaling cascade does exist, all its steps must be enzymatically catalyzed, as is evident for the classic cascade regulation of glycogen metabolism.
Other aspect worth brief discussion relevant to physiological significance of the mitochondrial ROS production is its dependence on oxygen concentration. In contrast to the cytochrome oxidase reaction, which has an apparent Km for oxygen significantly lower than its physiologically conceivable concentration (about 5-fold less than that in air-saturated aqueous solutions at normal pressure [228]), the specific activities of both “major” mitochondrial ROS generators (complex I [43, 131, 148, 149] and DLDH [146]) are simply proportional to oxygen concentration. Hydrogen peroxide formation by intact mitochondria is also a first-order process even under hyperoxic conditions [43]. Hyperbolic or other complex kinetics of the mitochondrial ROS production might be expected if it does have physiological function. On the other hand, simple proportionality of ROS formation to oxygen concentration may be used as an ideal oxygen sensing mechanism. Mammals have an extremely complex and well-developed hypoxia sensing system [229, 230], whereas the quantitatively major oxygen consumer, the mitochondrial respiratory chain, is not sensitive to oxygen concentration down to its very low level. The activities of ROS-producing enzymes may function as the primary sensors of oxygen. Interestingly, a special oxygen-sensing system exists – the α-oxoglutarate-dependent proline hydroxylase reaction participating in formation of so-called HIF (Hypoxia-Induced Factor) [231, 232].
The widespread opinion that mitochondria are the major source of ROS is apparently based on the fact that mitochondria are, indeed, the major consumers of oxygen. This can be exemplified by citing D. Harman, who wrote: “Mitochondria would be expected to be particularly subject to free radical induced change as over 90% of oxygen utilization by mammals takes place in them” [8]. We disagree with that and other similar statements. Mitochondrial respiration inevitably decreases, not increases, local concentration of oxygen in the vicinity of the centers potentially capable of ROS production. This would result in a decrease in ROS generation because of its first-order dependence on oxygen. Unfortunately, an extremely important general problem of valid quantitative determination of the intracellular oxygen concentration is not yet solved.
The rates of ROS production in mitochondria under “optimal” conditions correspond to only a very small percentage of their normal oxidase activity (~5 nmol/min per mg of protein). One milligram of the mitochondrial protein corresponds to approximately 1 µl of matrix water [233, 234]. If we consider the inner mitochondrial membrane as not permeable or poorly permeable to hydrogen peroxide, the specific activity of ROS-producing enzymes would result in a formation of 5 mM hydrogen peroxide after 1 min of normal respiration! Certainly, permeability for hydrogen peroxide and the presence of peroxidases and catalase are not accounted for in such an approximation. Nevertheless, possible gradients of hydrogen peroxide concentration between the mitochondrial matrix and cytoplasm cannot be neglected. A quantitative comparison of intact and permeabilized mitochondria suggests that all major activities (substrate oxidation and coupled ATP synthesis) are strictly limited (controlled) by the specific carriers [37].
ROS-producing activities of complex I and DLDH, the major generators, depend on the matrix redox potential, i.e. NAD+/NADH ratio [146]. The concentrations of free pyridine nucleotides in the mitochondrial matrix are not known. Their total amount in heart mitochondria can be approximated as 4-7 nmol per mg of protein [235-240], values corresponding to 4-7 mM concentration. It could thus be assumed that nucleotide-binding sites of complex I and DLDH are saturated. However, many NAD+-dependent dehydrogenases are present in the mitochondrial matrix, thus making determination of true free concentrations of nucleotides an unsolved problem important for general understanding of mitochondrial metabolism. The intramitochondrial NAD+/NADH ratio is determined by their functional state, as demonstrated in classical papers by Chance [154] and Klingenberg [238]. The physiologically relevant NAD+/NADH ratio in liver mitochondria was approximated as about 8 [239]. In isolated cardiomyocytes respiring on 10 mM glucose [241] or in heart perfused by 10 mM glucose, this ratio is closed to 1 [242]. Thus, apparent redox potential in the matrix is significantly higher than that conventionally used in model experiments on ROS production. It appears that in vivo mitochondrial ROS production is many-fold lower than measured under “optimal” conditions. In recent years, the term “Δp-dependent ROS-production” is frequently used. This assumes that membrane energization and de-energization increases and decreases ROS production, respectively. We believe that this terminology may be misinterpreted by a reader only superficially familiar with mitochondrial bioenergetics. The production of ROS depends on the amount (concentration) of the reactive sites and their oxygen accessibility. The steady-state NADH/NAD+ ratio, indeed, is decreased when respiration is activated by ADP (small drop of Δp) or by uncoupler (complete dissipation of Δp). The same is true for the ubisemiquinone/quinone couple, the most likely producer of ROS by complex III. It is important to understand that Δp itself does not affect the generation rate, rather it influences redox state of the oxygen reactive sites. The inhibitory effects of ADP and uncouplers on ROS production were demonstrated by Chance et al. as early as 1956 [48]. No correlation between membrane energization and ROS production exists; for example, rotenone and/or antimycin A stimulate mitochondrial ROS production, whereas either inhibitor completely de-energizes membranes. It may appear that this note is a matter of semantics; however, the expression “energy-dependent ROS generation” may easily lead to misinterpretation of the experimental data. Redox-dependent ROS production seems to more appropriately describe the real events.
There is no doubt that a number of enzymes, particularly those located in mitochondria, do produce hydrogen peroxide directly or via intermediately formed superoxide. The enzymes utilizing superoxide and hydrogen peroxide are frequently considered as an “antioxidant defense system”, and generation of ROS as an evolutionarily unfavorable leakage reaction (except for NAD(P)H oxidases, which perform a clearly defined function). An alternative view is that hydrogen peroxide is a normal metabolic intermediate that fulfills some important not yet clear functions. If a “leakage” hypothesis and “antioxidant defense system” are to be accepted, many more or less experimentally justified efforts to improve the defense (antioxidant drugs, particularly those mitochondrially targeted [243-245]) are certainly warrant. On the other hand, if the alternative view is correct the use of so-called antioxidant drugs may result in unfavorable consequences.
When discussing physiological and pathophysiological “oxidative stress”, one should keep in mind possible metabolic effects of “reductive stress” [246, 247]. Obvious consequences of “overreduction” would be: 1) abnormal anabolic activity including malignant growth; 2) an increase in fatty acid and fat biosynthesis; 3) disorder of amino acid transport systems (participation of glutathione in the γ-glutamate cycle [248]); 4) a decrease in normal insulin level due to an increase in the insulin:glutathione transhydrogenase reaction [249, 250]. Last, not the least consequence of reductive stress is a possible paradoxical activation of ROS production. As emphasized, the activities of all known ROS generators are sensitive to and determined by the NAD(P)+/NAD(P)H ratio. A decrease in the cellular redox potential (overreduction of pyridine nucleotides) would inevitably result in increase in ROS production.
CONCLUDING REMARKS
In this review we have described phenomenological characteristics of mitochondrial ROS production and some thoughts concerning its physiological significance. What are the most important topics for further studies in this field?
The search for reliable quantitative information about the rates of ROS production, decomposition, and steady-state concentration for isolated enzymes and for such a complex system as mitochondria or cells are greatly hampered by the absence of simple methods for their assay. Analytical biochemistry of ROS seems to be important area needing further development.
Some thoughts about reductive stress make promising the search for new specific or moonlighting ROS generators. It is expected, for example, that some classical participants of the antioxidant defense system such as glutathione reductase, similarly to its close relative DLDH, would be able to produce ROS under low redox potential conditions.
Despite countless published papers that suggest explicitly or implicitly the signaling functions of ROS, no direct evidence comparable to those established for true signaling molecules (cAMP, Ca2+, NO) have been obtained. Proof for this intriguing but still hypothetical function of ROS seems to be an important area of further studies in the field.
We are grateful to T. V. Zharova, A. V. Kareyeva, G. V. Gladyshev, and B. V. Chernyak for their most helpful comments.
This study was supported by grant 11-04-00916 to ADV and grant 12-04-00602 to VGG of the Russian Foundation for Basic Research and by NIH Fogarty International Research grant R03TW007825.
REFERENCES
1.Haldane, J. S., and Priestley, J. G. (1935)
Respiration, 2nd Edn., Oxford University Press.
2.Gerschman, R., Gilbert, D. L., Nye, S. W., Dwyer,
P., and Fenn, W. O. (1954) Science, 119, 623-626.
3.Harman, D. (1956) J. Gerontol., 11,
298-300.
4.Jensen, P. K. (1966) Biochim. Biophys. Acta,
122, 157-166.
5.McCord, J. M., and Fridovich, I. (1969) J. Biol.
Chem., 244, 6049-6055.
6.Keele, B. B., Jr., McCord, J. M., and Fridovich, I.
(1970) J. Biol. Chem., 245, 6176-6181.
7.Loschen, G., Flohe, L., and Chance, B. (1971)
FEBS Lett., 18, 261-264.
8.Harman, D. (1972) J. Am. Geriatr. Soc.,
20, 145-147.
9.Boveris, A., Oshino, N., and Chance, B. (1972)
Biochem. J., 128, 617-630.
10.Babior, B. M., Kipnes, R. S., and Curnutte, J. T.
(1973) J. Clin. Invest., 52, 741-744.
11.White, A. A., Crawford, K. M., Patt, C. S., and
Lad, P. J. (1976) J. Biol. Chem., 251, 7304-7312.
12.Veal, E. A., Day, A. M., and Morgan, B. A. (2007)
Mol. Cell, 26, 1-14.
13.Santos, C. X., Anilkumar, N., Zhang, M., Brewer,
A. C., and Shah, A. M. (2011) Free Radic. Biol. Med., 50,
777-793.
14.Imlay, J. A. (2003) Annu. Rev. Microbiol.,
57, 395-418.
15.Miller, A. F. (2012) FEBS Lett.,
586, 585-595.
16.Chen, Q., Vazquez, E. J., Moghaddas, S., Hoppel,
C. L., and Lesnefsky, E. J. (2003) J. Biol. Chem., 278,
36027-36031.
17.Turrens, J. F. (2003) J. Physiol.,
15, 335-344.
18.Staniek, K., and Nohl, H. (2000) Biochim.
Biophys. Acta, 1460, 268-275.
19.Brown, G. C., and Borutaite, V. (2012)
Mitochondrion, 12, 1-4.
20.George, P. (1965) The Fitness of Oxygen
(King, T. E., Mason, H. S., and Morrison, M., eds.) John Wiley &
Sons, Inc., New York-London-Sidney, pp. 3-36.
21.Paulus, A., Rossius, S. G., Dijk, M., and de
Vries, S. (2012) J. Biol. Chem., 287, 8830-8888.
22.Knowles, P. F., Gibson, J. F., Pick, F. M., and
Bray, R. C. (1969) Biochem. J., 111, 53-58.
23.Fridovich, I. (1974) Superoxide Dismutases.
Adv. Enzymol., 41, 35-97.
24.Clark, W. M. (1960) Oxidation-Reduction
Potential of Organic Systems, The Williams & Wilkins Co.,
Baltimore.
25.Sawyer, D. T., and Nanni, E. J., Jr. (1981) in
Oxygen and Oxy-Radicals in Chemistry and Biology (Rodgers, A.
J., and Powers, E. L., eds.) Academic Press, New York-London-Sydney-San
Francisco, pp. 15-44.
26.Wood, P. M. (1988) Biochem. J.,
253, 287-289.
27.Massey, V. (1994) J. Biol. Chem.,
269, 22459-22462.
28.Bakeeva, L. E., Chentsov, Y. S., Jasaitis, A. A.,
and Skulachev, V. P. (1972) Biochim. Biophys. Acta, 275,
319-332.
29.Amchenkova, A. A., Bakeeva, L. E., Chentsov, Y.
S., Skulachev, V. P., and Zorov, D. B. (1988) J. Cell. Biol.,
107, 481-495.
30.Bing, R. J., Siegel, A., Vitale, A., Balboni, F.,
Sparks, E., Taeschler, M., Klapper, M., and Edwards, S. (1953) Am.
J. Med., 15, 284-296.
31.Bing, R. J., Siegel, A., Ungar, I., and Gilbert,
M. (1954) Am. J. Med., 16, 504-515.
32.Chance, B., and Williams, G. R. (1955) J.
Biol. Chem., 217, 409-427.
33.Takahashi, M. A., and Asada, K. (1983) Arch.
Biochem. Biophys., 226, 558-566.
34.Gus’kova, R. A., Ivanov, I. I.,
Kol’tover, V. K., Akhobadze, V. V., and Rubin, A. B. (1984)
Biochim. Biophys. Acta, 778, 579-585.
35.Grivennikova, V. G., Kareyeva, A. V., and
Vinogradov, A. D. (2010) Biochim. Biophys. Acta, 1797,
939-944.
36.Grivennikova, V. G., Kapustin, A. N., and
Vinogradov, A. D. (2001) J. Biol. Chem., 276,
9038-9044.
37.Gostimskaya, I. S., Grivennikova, V. G., Zharova,
T. V., Bakeeva, L. E., and Vinogradov, A. D. (2003) Anal.
Biochem., 313, 46-52.
38.Kotlyar, A. B., and Vinogradov, A. D. (1990)
Biochim. Biophys. Acta, 1019, 151-158.
39.Tarpey, M. M., and Fridovich, I. (2001) Circ.
Res., 89, 224-236.
40.Miwa, S., and Brand, M. D. (2005) Biochim.
Biophys. Acta, 1709, 214-219.
41.Chance, B., Sies, H., and Boveris, A. (1979)
Physiol. Rev., 59, 527-605.
42.Zhou, M., Diwu, Z., Panchuk-Voloshina, N., and
Haugland, R. P. (1997) Anal. Biochem., 253, 162-168.
43.Boveris, A., and Chance, B. (1973) Biochem.
J., 134, 707-716.
44.Tahara, E. B., Navarete, F. D., and Kowaltowski,
A. J. (2009) Free Radic. Biol. Med., 46, 1283-1297.
45.Lambert, A. J., Boysen, H. M., Buckingham, J. A.,
Yang, T., Podlutsky, A., Austad, S. N., Kunz, T. H., Buffenstein, R.,
and Brand, M. D. (2007) Aging Cell, 6, 607-168.
46.St-Pierre, J., Buckingham, J. A., Roebuck, S. J.,
and Brand, M. D. (2002) J. Biol. Chem., 277,
44784-44790.
47.Korshunov, S. S., Skulachev, V. P., and Starkov,
A. A. (1997) FEBS Lett., 416, 15-18.
48.Chance, B., and Williams, G. R. (1956) Adv.
Enzymol. Relat. Subj. Biochem., 17, 65-134.
49.Hinkle, P. C., Butow, R. A., Racker, E., and
Chance, B. (1967) J. Biol. Chem., 242, 5169-5173.
50.Takeshige, K., and Minakami, S. (1979)
Biochem. J., 180, 129-135.
51.Turrens, J. F., and Boveris, A. (1980)
Biochem. J., 191, 421-427.
52.Krishnamoorthy, G., and Hinkle, P. C. (1988)
J. Biol. Chem., 263, 17566-17575.
53.Ksenzenko, M., Konstantinov, A. A., Khomutov, G.
B., Tikhonov, A. N., and Ruuge, E. K. (1983) FEBS Lett.,
155, 19-24.
54.Sun, J., and Trumpower, B. L. (2003) Arch.
Biochem. Biophys., 419, 198-206.
55.Loschen, G., Azzi, A., Richter, C., and Flohe, L.
(1974) FEBS Lett., 42, 68-72.
56.Brandt, U. (2006) Annu. Rev. Biochem.,
75, 69-92.
57.Cecchini, G. (2003) Annu. Rev. Biochem.,
72, 77-109.
58.Hunte, C., Solmaz, S., Palsdottir, H., and Wenz,
T. (2008) Results Probl. Cell Differ., 45, 253-278.
59.Carothers, D. J., Pons, G., and Patel, M. S.
(1989) Arch. Biochem. Biophys., 268, 409-425.
60.Bedard, K., and Krause, K. H. (2007) Physiol.
Rev., 87, 245-313.
61.Edmondson, D. E., Binda, C., and Mattevi, A.
(2004) Neurotoxicology, 25, 63-72.
62.Watmough, N. J., and Frerman, F. E. (2004)
Biochim. Biophys. Acta, 1797, 1910-1916.
63.Mracek, T., Drahota, Z., and Houstek, J. (2013)
Biochim. Biophys. Acta, 1827, 401-410.
64.Evans, D. R., and Guy, H. I. (2004) J. Biol.
Chem., 279, 33035-33038.
65.Miller, R. W., Kerr, C. T., and Curry, J. R.
(1968) Can. J. Biochem., 46, 1099-1106.
66.Kennedy, J. (1973) Arch. Biochem.
Biophys., 157, 369-373.
67.Chen, J. J., and Jones, M. E. (1976) Arch.
Biochem. Biophys., 176, 82-90.
68.Angermuller, S., and Loffler, M. (1995)
Histochem. Cell. Biol., 103, 287-292.
69.Loffler, M., Becker, C., Wegerle, E., and
Schuster, G. (1996) Histochem. Cell. Biol., 105,
119-128.
70.Rawls, J., Knecht, W., Diekert, K., Lill, R., and
Loffler, M. (2000) Eur. J. Biochem., 267, 2079-2087.
71.Norager, S., Jensen, K. F., Bjornberg, O., and
Larsen, S. (2002) Structure, 10, 1211-1223.
72.Forman, H. J., and Kennedy, J. (1975) J. Biol.
Chem., 250, 4322-4326.
73.Forman, H. J., and Kennedy, J. (1976) Arch.
Biochem. Biophys., 173, 219-224.
74.Hail, N., Jr., Chen, P., Kepa, J. J., Bushman, L.
R., and Shearn, C. (2010) Free Radic. Biol. Med., 49,
109-116.
75.Klingenberg, M. (1970) Eur. J. Biochem.,
13, 247-252.
76.Mracek, T., Pecinova, A., Vrbacky, M., Drahota,
Z., and Houstek, J. (2009) Arch. Biochem. Biophys., 481,
30-36.
77.Orr, A. L., Quinlan, C. L., Perevoshchikova, I.
V., and Brand, M. D. (2012) J. Biol. Chem., 287,
42921-42935.
78.Dummler, K., Muller, S., and Seitz, H. J. (1996)
Biochem. J., 317, 913-918.
79.Brown, L. J., MacDonald, M. J., Lehn, D. A., and
Moran, S. M. (1994) J. Biol. Chem., 269, 14363-14366.
80.Yeh, J. I., Chinte, U., and Du, S. (2008)
Proc. Natl. Acad. Sci. USA, 105, 3280-3285.
81.Drahota, Z., Chowdhury, S. K., Floryk, D.,
Mracek, T., Wilhelm, J., Rauchova, H., Lenaz, G., and Houstek, J.
(2002) J. Bioenerg. Biomembr., 34, 105-113.
82.Chen, F., Haigh, S., Barman, S., and Fulton, D.
J. (2012) Front. Physiol., 3, 1-12.
83.Takac, I., Schroder, K., Zhang, L., Lardy, B.,
Anilkumar, N., Lambeth, J. D., Shah, A. M., Morel, F., and Brandes, R.
P. (2011) J. Biol. Chem., 286, 13304-13313.
84.Geiszt, M., Kopp, J. B., Varnai, P., and Leto, T.
L. (2000) Proc. Natl. Acad. Sci. USA, 97, 8010-8014.
85.Kuroda, J., Ago, T., Matsushima, S., Zhai, P.,
Schneider, M. D., and Sadoshima, J. (2010) Proc. Natl. Acad. Sci.
USA, 107, 15565-15570.
86.Ago, T., Kuroda, J., Pain, J., Fu, C., Li, H.,
and Sadoshima, J. (2010) Circ. Res., 106, 1253-1264.
87.Maejima, Y., Kuroda, J., Matsushima, S., Ago, T.,
and Sadoshima, J. (2011) J. Mol. Cell. Cardiol., 50,
408-416.
88.Schnaitman, C., Erwin, V. G., and Greenawalt, J.
W. (1967) J. Cell. Biol., 32, 719-735.
89.Binda, C., Newton-Vinson, P., Hubalek, F.,
Edmondson, D. E., and Mattevi, A. (2002) Nat. Struct. Biol.,
9, 22-26.
90.De Colibus, L., Li, M., Binda, C., Lustig, A.,
Edmondson, D. E., and Mattevi, A. (2005) Proc. Natl. Acad. Sci.
USA, 102, 12684-12689.
91.Edmondson, D. E., Binda, C., Wang, J., Upadhyay,
A. K., and Mattevi, A. (2009) Biochemistry, 48,
4220-4230.
92.Sivasubramaniam, S. D., Finch, C. C., Rodriguez,
M. J., Mahy, N., and Billett, E. E. (2003) Cell. Tissue Res.,
313, 291-300.
93.Kaludercic, N., Carpi, A., Menabo, R., Di Lisa,
F., and Paolocci, N. (2011) Biochim. Biophys. Acta, 1813,
1323-1332.
94.Bianchi, P., Kunduzova, O., Masini, E., Cambon,
C., Bani, D., Raimondi, L., Seguelas, M. H., Nistri, S., Colucci, W.,
Leducq, N., and Parini, A. (2005) Circulation, 112,
3297-3305.
95.Maurel, A., Hernandez, C., Kunduzova, O.,
Bompart, G., Cambon, C., Parini, A., and Frances, B. (2003) Am. J.
Physiol. Heart Circ. Physiol., 284, H1460-H1467.
96.Crane, F. L., and Beinert, H. (1956) J. Biol.
Chem., 218, 717-731.
97.Kim, J. J., and Miura, R. (2004) Eur. J.
Biochem., 271, 483-493.
98.Roberts, D. L., Frerman, F. E., and Kim, J.-J.
(1996) Proc. Natl. Acad. Sci. USA, 93, 14355-14360.
99.Ramsay, R. R., Steenkamp, D. J., and Husain, M.
(1987) Biochem. J., 241, 883-892.
100.Ruzicka, F. J., and Beinert, H. (1977) J.
Biol. Chem., 252, 8440-8445.
101.Zhang, J., Frerman, F. E., and Kim, J.-J.
(2006) Proc. Natl. Acad. Sci. USA, 103, 16212-16217.
102.Seifert, E. L., Estey, C., Xuan, J. Y., and
Harper, M. E. (2010) J. Biol. Chem., 285, 5748-5758.
103.Schonfeld, P., and Wojtczak, L. (2012)
Biochim. Biophys. Acta, 1817, 410-418.
104.Rosca, M. G., Vazquez, E. J., Chen, Q., Kerner,
J., Kern, T. S., and Hoppel, C. L. (2012) Diabetes, 61,
2074-2083.
105.Wang, R., and Thorpe, C. (1991)
Biochemistry, 30, 7895-7901.
106.Carroll, J., Fearnley, I. M., Skehel, J. M.,
Shannon, R. J., Hirst, J., and Walker, J. E. (2006) J. Biol.
Chem., 281, 32724-32727.
107.Balsa, E., Marco, R., Perales-Clemente, E.,
Szklarczyk, R., Calvo, E., Landazuri, M. O., and Enriquez, J. A. (2012)
Cell Metab., 16, 378-386.
108.Morgner, N., Zickermann, V., Kerscher, S.,
Wittig, I., Abdrakhmanova, A., Barth, H. D., Brutschy, B., and Brandt,
U. (2008) Biochim. Biophys. Acta, 1777, 1384-1391.
109.Drose, S., Krack, S., Sokolova, L., Zwicker,
K., Barth, H. D., Morgner, N., Heide, H., Steger, M., Nubel, E.,
Zickermann, V., Kerscher, S., Brutschy, B., Radermacher, M., and
Brandt, U. (2011) PLoS Biol., 9, e1001128, 1-11.
110.Walker, J. E. (1992) Q. Rev. Biophys.,
25, 253-324.
111.Yagi, T., and Matsuno-Yagi, A. (2003)
Biochemistry, 42, 2266-2274.
112.Rao, N. A., Felton, S. P., Huennekens, F. M.,
and Mackler, B. (1963) J. Biol. Chem., 238, 449-455.
113.Ohnishi, T., Sled, V. D., Yano, T., Yagi, T.,
Burbaev, D. S., and Vinogradov, A. D. (1998) Biochim. Biophys.
Acta, 1365, 301-308.
114.Sled, V. D., Friedrich, T., Leif, H., Weiss,
H., Meinhardt, S. W., Fukumori, Y., Calhoun, M. W., Gennis, R. B., and
Ohnishi, T. (1993) J. Bioenerg. Biomembr., 25,
347-356.
115.Ohnishi, T., and Salerno, J. C. (1982)
Iron-Sulfur Proteins, Vol. IV (Spiro, T., ed.) Wiley Publishing
Co. Inc., N. Y., pp. 285-327.
116.Beinert, H., and Albracht, S. P. J. (1982)
Biochim. Biophys. Acta, 683, 245-277.
117.Vinogradov, A. D., Sled, V. D., Burbaev, D. S.,
Grivennikova, V. G., Moroz, I. A., and Ohnishi, T. (1995) FEBS
Lett., 370, 83-87.
118.Sazanov, L. A. (2007) Biochemistry,
46, 2275-2288.
119.Efremov, R. G., Baradaran, R., and Sazanov, L.
A. (2010) Nature, 465, 441-445.
120.Clason, T., Ruiz, T., Schagger, H., Peng, G.,
Zickermann, V., Brandt, U., Michel, H., and Radermacher, M. (2010)
J. Struct. Biol., 169, 81-88.
121.Sazanov, L. A., and Hinchliffe, P. (2006)
Science, 311, 1430-1436.
122.Efremov, R. G., and Sazanov, L. A. (2011)
Nature, 476, 414-420.
123.Hunte, C., Zickermann, V., and Brandt, U.
(2010) Science, 329, 448-451.
124.Ohnishi, T. (1975) Biochim. Biophys.
Acta, 387, 475-490.
125.Mathiesen, C., and Hagerhall, C. (2002)
Biochim. Biophys. Acta, 1556, 121-132.
126.Baradaran, R., Berrisford, J. M., Minhas, G.
S., and Sazanov, L. A. (2013) Nature, 494, 443-448.
127.Grivennikova, V. G., and Vinogradov, A. D.
(2013) Biochim. Biophys. Acta, 1827, 446-454.
128.Votyakova, T. V., and Reynolds, I. J. (2001)
J. Neurochem., 79, 266-277.
129.Grivennikova, V. G., and Vinogradov, A. D.
(2006) Biochim. Biophys. Acta, 1757, 553-561.
130.Kang, D., Narabayashi, H., Sata, T., and
Takeshige, K. (1983) J. Biochem., 94, 1301-1306.
131.Kussmaul, L., and Hirst, J. (2006) Proc.
Natl. Acad. Sci. USA, 103, 7607-7612.
132.Esterhazy, D., King, M. S., Yakovlev, G., and
Hirst, J. (2008) Biochemistry, 47, 3964-3971.
133.Pepelina, T. Y., Chertkova, R. V.,
Ostroverkhova, T. V., Dolgikh, D. A., Kirpichnikov, M. P.,
Grivennikova, V. G., and Vinogradov, A. D. (2009) Biochemistry
(Moscow), 74, 625-632.
134.Grivennikova, V. G., Ushakova, A. V., Cecchini,
G., and Vinogradov, A. D. (2003) FEBS Lett., 549,
39-42.
135.Vinogradov, A. D. (2008) Biochim. Biophys.
Acta, 1777, 729-734.
136.Kotlyar, A. B., Karliner, J. S., and Cecchini,
G. (2005) FEBS Lett., 579, 4861-4866.
137.Grivennikova, V. G., Kotlyar, A. B., Karliner,
J. S., Cecchini, G., and Vinogradov, A. D. (2007) Biochemistry,
46, 10971-10978.
138.Zharova, T. V., and Vinogradov, A. D. (1997)
Biochim. Biophys. Acta, 1320, 256-264.
139.Ragan, C. I., and Bloxham, D. P. (1977)
Biochem. J., 163, 605-615.
140.Majander, A., Finel, M., and Wikstrom, M.
(1994) J. Biol. Chem., 269, 21037-21042.
141.Degli Esposti, M. (1998) Biochim. Biophys.
Acta, 1364, 222-235.
142.Miyoshi, H. (1998) Biochim. Biophys.
Acta, 1364, 236-244.
143.Okun, J. G., Lummen, P., and Brandt, U. (1999)
J. Biol. Chem., 274, 2625-2630.
144.Kushnareva, Y., Murphy, A. N., and Andreyev, A.
(2002) Biochem. J., 368, 545-553.
145.Kudin, A. P., Bimpong-Buta, N. Y., Vielhaber,
S., Elger, C. E., and Kunz, W. S. (2004) J. Biol. Chem.,
279, 4127-4135.
146.Kareyeva, A. V., Grivennikova, V. G., and
Vinogradov, A. D. (2012) Biochim. Biophys. Acta, 1817,
1879-1885.
147.Sled, V. D., Rudnitzky, N. I., Hatefi, Y., and
Ohnishi, T. (1994) Biochemistry, 33, 10069-10075.
148.Turrens, J. F., Freeman, B. A., Levitt, J. G.,
and Crapo, J. D. (1982) Arch. Biochem. Biophys., 217,
401-410.
149.Vinogradov, A. D., and Grivennikova, V. G.
(2005) Biochemistry (Moscow), 70, 120-127.
150.Ohnishi, T. (1998) Biochim. Biophys.
Acta, 1364, 186-206.
151.Galkin, A., and Brandt, U. (2005) J. Biol.
Chem., 280, 30129-30135.
152.Treberg, J. R., Quinlan, C. L., and Brand, M.
D. (2011) J. Biol. Chem., 286, 27103-27110.
153.Ohnishi, S. T., Shinzawa-Itoh, K., Ohta, K.,
Yoshikawa, S., and Ohnishi, T. (2010) Biochim. Biophys. Acta,
1797, 1901-1909.
154.Chance B., and Hollunger, G. (1961) J. Biol.
Chem., 236, 1555-1561.
155.Schofield, C. J., and Zhang, Z. (1999) Curr.
Opin. Struct. Biol., 9, 722-731.
156.Epstein, A. C., Gleadle, J. M., McNeill, L. A.,
Hewitson, K. S., O’Rourke, J., Mole, D. R., Mukherji, M., Metzen,
E., Wilson, M. I., Dhanda, A., Tian, Y. M., Masson, N., Hamilton, D.
L., Jaakkola, P., Barstead, R., Hodgkin, J., Maxwell, P. H., Pugh, C.
W., Schofield, C. J., and Ratcliffe, P. J. (2001) Cell,
107, 43-54.
157.Nguyen, E., and Picklo, M. J. Sr. (2003)
Biochim. Biophys. Acta, 1637, 107-112.
158.Maklashina, E. O., Sled, V. D., and Vinogradov,
A. D. (1994) Biochemistry (Moscow), 59, 707-713.
159.Maklashina, E. O., and Vinogradov, A. D. (1994)
Biochemistry (Moscow), 59, 1221-1226.
160.Vinogradov, A. D. (1998) Biochim. Biophys.
Acta, 1364, 169-185.
161.Vinogradov, A. D., and Grivennikova, V. G.
(2001) IUBMB Life, 52, 129-134.
162.Maklashina, E. O., Sher, Y., Zhou, H.-Z., Gray,
M. O., Karliner, J. S., and Cecchini, G. (2002) Biochim. Biophys.
Acta, 1556, 6-12.
163.Loskovich, M. V., Grivennikova, V. G.,
Cecchini, G., and Vinogradov, A. D. (2005) Biochem. J.,
387, 677-683.
164.Kotlyar, A. B., Sled, V. D., and Vinogradov, A.
D. (1992) Biochim. Biophys. Acta, 1098, 144-150.
165.Kalashnikov, D. S., Grivennikova, V. G., and
Vinogradov, A. D. (2011) Biochemistry (Moscow), 76,
968-975.
166.Galkin, A., Meyer, B., Wittig, I., Karas, M.,
Schagger, H., Vinogradov, A., and Brandt, U. (2008) J. Biol.
Chem., 283, 20907-20913.
167.Gavrikova, E. V., and Vinogradov, A. D. (1999)
FEBS Lett., 455, 36-40.
168.Gostimskaya, I. S., Cecchini, G., and
Vinogradov, A. D. (2006) Biochim. Biophys. Acta, 1757,
1155-1161.
169.Galkin, A., and Moncada, S. (2007) J. Biol.
Chem., 282, 37448-37453.
170.Chouchani, E. T., Methner, C., Nadtochiy, S.
M., Logan, A., Pell, V. R., Ding, S., James, A. M., Cocheme, H. M.,
Reinhold, J., Lilley, K. S., Partridge, L., Fearnley, I. M., Robinson,
A. J., Hartley, R. C., Smith, R. A., Krieg, T., Brookes, P. S., and
Murphy, M. P. (2013) Nat. Med., 19, 753-759.
171.Yankovskaya, V., Horsefield, R., Tornroth, S.,
Luna-Chavez, C., Miyoshi, H., Leger, C., Byrne, B., Cecchini, G., and
Iwata, S. (2003) Science, 299, 700-704.
172.Zhang, L., Yu, L., and Yu, C. A. (1998) J.
Biol. Chem., 273, 33972-33976.
173.Messner, K. R., and Imlay, J. A. (2002) J.
Biol. Chem., 277, 42563-42571.
174.Quinlan, C. L., Orr, A. L., Perevoshchikova, I.
V., Treberg, J. R., Ackrell, B. A., and Brand, M. D. (2012) J. Biol.
Chem., 287, 27255-27264.
175.Moreno-Sanchez, R., Hernandez-Esquivel, L.,
Rivero-Segura, N. A., Marin-Hernandez, A., Neuzil, J., Ralph, S. J.,
and Rodriguez-Enriquez, S. (2013) FEBS J., 280,
927-938.
176.Berry, E. A., Huang, L. S., Saechao, L. K.,
Pon, N. G., Valkova-Valchanova, M., and Daldal, F. (2004)
Photosynth. Res., 81, 251-275.
177.Kleinschroth, T., Castellani, M., Trinh, C. H.,
Morgner, N., Brutschy, B., Ludwig, B., and Hunte, C. (2011) Biochim.
Biophys. Acta, 1807, 1606-1615.
178.Xia, D., Yu, C. A., Kim, H., Xia, J. Z.,
Kachurin, A. M., Zhang, L., Yu, L., and Deisenhofer, J. (1997)
Science, 277, 60-66.
179.Zhang, Z., Huang, L., Shulmeister, V. M., Chi,
Y. I., Kim, K. K., Hung, L. W., Crofts, A. R., Berry, E. A., and Kim,
S. H. (1998) Nature, 392, 677-684.
180.Iwata, S., Lee, J. W., Okada, K., Lee, J. K.,
Iwata, M., Rasmussen, B., Link, T. A., Ramaswamy, S., and Jap, B. K.
(1998) Science, 281, 64-71.
181.Lange, C., and Hunte, C. (2002) Proc. Natl.
Acad. Sci. USA, 99, 2800-2805.
182.Hunte, C., Palsdottir, H., and Trumpower, B. L.
(2003) FEBS Lett., 545, 39-46.
183.Mitchell, P. (1975) FEBS Lett.,
56, 1-6.
184.Mitchell, P. (1975) FEBS Lett.,
59, 137-139.
185.Drose, S., and Brandt, U. (2012) Adv. Exp.
Med. Biol., 748, 145-169.
186.Crofts, A. R. (2004) Biochim. Biophys.
Acta, 1655, 77-92.
187.Brandt, U., and Trumpower, B. (1994) Crit.
Rev. Biochem. Mol. Biol., 29, 165-197.
188.Trumpower, B. L. (2002) Biochim. Biophys.
Acta, 1555, 166-173.
189.Osyczka, A., Moser, C. C., Daldal, F., and
Dutton, P. L. (2004) Nature, 427, 607-612.
190.Osyczka, A., Moser, C. C., and Dutton, P. L.
(2005) Trends Biochem. Sci., 30, 176-182.
191.Zhu, J., Egawa, T., Yeh, S. R., Yu, L., and Yu,
C. A. (2007) Proc. Natl. Acad. Sci. USA, 104,
4864-4869.
192.Brandt, U. (1998) Biochim. Biophys.
Acta, 1365, 261-268.
193.Covian, R., and Trumpower, B. L. (2008)
Biochim. Biophys. Acta, 1777, 1044-1052.
194.Covian, R., and Trumpower, B. L. (2008)
Biochim. Biophys. Acta, 1777, 1079-1091.
195.Castellani, M., Covian, R., Kleinschroth, T.,
Anderka, O., Ludwig, B., and Trumpower, B. L. (2010) J. Biol.
Chem., 285, 502-510.
196.Bleier, L., and Drose, S. (2013) Biochim.
Biophys. Acta, http://dx.doi.org/10.1016/j.bbabio.2012.12.002.
197.Lanciano, P., Khalfaoui-Hassani, B., Selamoglu,
N., Ghelli, A., Rugolo, M., and Daldal, F. (2013) Biochim. Biophys.
Acta, http://dx.doi.org/10.1016/j.bbabio.2013.03.009.
198.Yin, Y., Yang, S., Yu, L., and Yu, C. A. (2010)
J. Biol. Chem., 285, 17038-17045.
199.Borek, A., Sarewicz, M., and Osyczka, A. (2008)
Biochemistry, 47, 12365-12370.
200.Sarewicz, M., Borek, A., Cieluch, E.,
Swierczek, M., and Osyczka, A. (2010) Biochim. Biophys. Acta,
1797, 1820-1827.
201.Rottenberg, H., Covian, R., and Trumpower, B.
L. (2009) J. Biol. Chem., 284, 19203-19210.
202.Drose, S., and Brandt, U. (2008) J. Biol.
Chem., 283, 21649-21654.
203.Zhou, F., Yin, Y., Su, T., Yu, L., and Yu, C.
A. (2013) Biochim. Biophys. Acta, 1817, 2103-2109.
204.Muller, F. L., Liu, Y., and Van Remmen, H.
(2004) J. Biol. Chem., 279, 49064-49073.
205.Voet, D., and Voet, J. G. (1990)
Biochemistry, John Wiley & Sons, N. Y., pp. 382-387.
206.Babady, N. E., Pang, Y. P., Elpeleg, O., and
Isaya, G. (2007) Proc. Natl. Acad. Sci. USA, 104,
6158-6163.
207.Xia, L., Bjornstedt, M., Nordman, T., Eriksson,
L. C., and Olsson, J. M. (2001) Eur. J. Biochem., 268,
1486-1490.
208.Tyagi, T. K., Ponnan, P., Singh, P., Bansal,
S., Batra, A., Collin F., Guillonneau, F., Jore, D., Patkar, S. A.,
Saxena, R. K., Parnar, V. S., Rastogi, R. C., and Raj, H. G. (2009)
Biochimie, 91, 868-875.
209.Kareyeva, A. V., Grivennikova, V. G., Cecchini,
G., and Vinogradov, A. D. (2011) FEBS Lett., 585,
385-389.
210.Lester, J., and Reed, L. G. (2001) J. Biol.
Chem., 276, 38329-38336.
211.Yeaman, S. J. (1989) Biochem. J.,
257, 625-632.
212.Kikuchi, G. (1973) Mol. Cell. Biochem.,
1, 169-187.
213.Chandrasekhar, K., Wang, J., Arjunan, P., Sax,
M., Park, Y. H., Nemeria, N. S., Kumaran, S., Song, J., Jordan, F., and
Furey, W. (2013) J. Biol. Chem., 288, 15402-15417.
214.Kenney, W. C., Zakim, D., Hogue, P. K., and
Singer, T. P. (1972) Eur. J. Biochem., 28, 253-260.
215.Danson, M. J., Eisenthal, R., Hall, S.,
Kessell, S. R., and Williams, D. L. (1984) Biochem. J.,
218, 811-818.
216.Danson, M. J., Conroy, K., McQuattie, A., and
Stevenson, K. J. (1987) Biochem. J., 243, 661-665.
217.Brautigam, C. A., Chuang, J. L., Tomchick, D.
R., Machius, M., and Chuang, D. T. (2005) J. Mol. Biol.,
350, 543-552.
218.Matthews, R. G., Ballou, D. P., Thorpe, C., and
Williams, C. H., Jr. (1977) J. Biol. Chem., 252,
3199-3207.
219.Ghisla, S., and Massey, V. (1989) Eur. J.
Biochem., 181, 1-17.
220.Starkov, A. A., Fiskum, G., Chinopoulos, C.,
Lorenzo, B. J., Browne, S. E., Patel, M. S., and Beal, M. F. (2004)
J. Neurosci., 24, 7779-7788.
221.Bunik, V. I., and Sievers, C. (2002) Eur. J.
Biochem., 269, 5004-5015.
222.Huennekens, F. M., Basford, R. E., and Gabrio,
B. W. (1955) J. Biol. Chem., 213, 951-967.
223.Tretter, L., and Adam-Vizi, V. (2004) J.
Neurosci., 24, 7771-7778.
224.Grivennikova, V. G., Cecchini, G., and
Vinogradov, A. D. (2008) FEBS Lett., 582, 2719-2724.
225.Adam-Vizi, V., and Tretter, L. (2013)
Neurochem. Int., 62, 757-763.
226.Tretter, L., and Adam-Vizi, V. (2000) J.
Neurosci., 20, 8972-8979.
227.Chinopoulos, C., Tretter, L., and Adam-Vizi, V.
(1999) J. Neurochem., 73, 220-228.
228.Wilson, D. F. (2008) Am. J. Physiol. Heart
Circ. Physiol., 294, H11-13.
229.Wenger, R. H. (2000) J. Exp. Biol.,
203, 1253-1263.
230.Kenneth, N. S., and Rocha, S. (2008)
Biochem. J., 414, 19-29.
231.Goda, N., and Kanai, M. (2012) Int. J.
Hematol., 95, 457-463.
232.Semenza, G. L. (2012) Cell, 148,
399-408.
233.Harris, E. J., and van Dam, K. (1968)
Biochem. J., 106, 759-766.
234.Halestrap, A. P., and Quinlan, P. T. (1983)
Biochem. J., 214, 387-393.
235.Glock, G. E., and McLean, P. (1956) Exp.
Cell. Res., 11, 234-236.
236.Jacobson, K. B., and Kaplan N. O. (1957) J.
Biol. Chem., 226, 603-613.
237.Lester, R. L., and Hatefi, Y. (1958)
Biochim. Biophys. Acta, 29, 103-112.
238.Klingenberg, M., Slenczka, W., and Ritt, E.
(1959) Biochem. Z., 332, 47-66.
239.Williamson, D. H., Lund, P., and Krebs, H. A.
(1967) Biochem. J., 103, 514-527.
240.Klingenberg, M. (1968) Biological
Oxidation (Singer, T. P., ed.) John Wiley Interscience Publisher,
N. Y., pp. 3-54.
241.Eng, J., Lynch, R. M., and Balaban, R. S.
(1989) Biophys. J., 55, 621-630.
242.Hassinen, I. E. (1986) Biochim. Biophys.
Acta, 853, 135-151.
243.Skulachev, V. P. (2011) Aging, 3,
1045-1050.
244.Skulachev, M. V., Antonenko, Y. N., Anisimov,
V. N., Chernyak, B. V., Cherepanov, D. A., Chistyakov, V. A., Egorov,
M. V., Kolosova, N. G., Korshunova, G. A., Lyamzaev, K. G., Plotnikov,
E. Y., Roginsky, V. A., Savchenko, A. Y., Severina, I. I., Severin, F.
F., Shkurat, T. P., Tashlitsky, V. N., Shidlovsky, K. M., Vyssokikh, M.
Y., Zamyatnin, A. A., Jr., Zorov, D. B., and Skulachev, V. P. (2011)
Curr. Drug Targets, 12, 800-826.
245.Murphy, M. P. (2013) Free Radic. Biol.
Med., http://dx.doi.org/10.1016/j.freeradbiomed.2013.04.010.
246.Kehrer, J. P. (2007) in Encyclopedia of
Stress (Fink, G., ed.) 2nd Edn., Academic Press, Oxford, Vol. 3,
pp. 331-335.
247.Zhang, H., Limphong, P., Pieper, J., Liu, Q.,
Rodesch, C. K., Christians, E., and Benjamin, I. J. (2012) FASEB
J., 26, 1442-1451.
248.Griffith, O. W., Bridges, R. J., and Meister,
A. (1979) Proc. Natl. Acad. Sci. USA, 76, 6319-6322.
249.Chandler, M. L., and Varandani, P. T. (1975)
Biochemistry, 14, 2107-2115.
250.Cordes, C. M., Bennett, R. G., Siford, G. L.,
and Hamel, F. G. (2011) PLoS One, 6, e18138.