REVIEW: Noncatalytic Nucleotide Binding Sites: Properties and Mechanism of Involvement in ATP Synthase Activity Regulation
A. N. Malyan
Institute of Basic Biological Problems, Russian Academy of Sciences, 142290 Pushchino, Moscow Region, Russia; E-mail: A_Malyan@issp.serpukhov.su; alexander.malyan@gmail.com
Received June 11, 2013
ATP synthases (FoF1-ATPases) of chloroplasts, mitochondria, and bacteria catalyze ATP synthesis or hydrolysis coupled with the transmembrane transfer of protons or sodium ions. Their activity is regulated through their reversible inactivation resulting from a decreased transmembrane potential difference. The inactivation is believed to conserve ATP previously synthesized under conditions of sufficient energy supply against unproductive hydrolysis. This review is focused on the mechanism of nucleotide-dependent regulation of the ATP synthase activity where the so-called noncatalytic nucleotide binding sites are involved. Properties of these sites varying upon free enzyme transition to its membrane-bound form, their dependence on membrane energization, and putative mechanisms of noncatalytic site-mediated regulation of the ATP synthase activity are discussed.
KEY WORDS: noncatalytic sites, F1-ATPase, ATP synthase, activation/inactivation, regulationDOI: 10.1134/S0006297913130099
Abbreviations: AMP-PNP, 5′-adenylyl-β,γ-imido diphosphate; FSBA, 5′-p-fluorosulfonylbenzoyladenosine.
In chloroplasts, mitochondria, and bacteria the final step of energy
transformation is performed by a special polypeptide complex termed ATP
synthase (FoF1-ATPase) through coupling of
transmembrane proton (sodium ion) transfer with phosphorylation of
adenosine diphosphate. In the course of evolution, to satisfy the
demand for adaptation of the energy machinery to varying conditions,
this complex acquired gene-encoded regulatory mechanisms responsible
for modulation of its catalytic activity. The principles of operation
of these mechanisms cannot be understood without knowledge of the
molecular structure of ATP synthase. To date, the best-studied
structures are those of mitochondrial and bacterial ATP synthases [1-5]. Studies on chloroplast ATP
synthase structure are much less advanced, which can be explained
mostly by difficulties of its crystallization. The situation is
improved by the fact that ATP synthase complexes of different genesis
have the same minimal subunit composition and are quite homologous to
one another, which determines similarity of their principle
functions.
The common feature of all ATP synthases is their reversible inactivation occurring at decreased transmembrane difference of proton potentials (energy-dependent regulation). This inactivation is believed to protect ATP previously synthesized in conditions of sufficient energy supply against unproductive hydrolysis. Both overall and specific regulatory mechanisms of various organelles are realized according to this type of regulation. The overall mechanisms are represented by nucleotide-dependent regulation of the ATPase activity of ATP synthases. The specific mechanisms differ in involvement of the ε-subunit (chloroplasts, bacteria), the IF1-subunit (mitochondria), and their sensitivity to ATP. In chloroplasts, there is a specific regulatory mechanism that depends on the stroma redox potential and is realized through the redox reaction between endogenous thioredoxin and the γ-subunit disulfide bond (thiol-dependent regulation) (for more details, see [6-9]). The ATP synthases contain three catalytic nucleotide-binding sites and three other sites termed as “noncatalytic” because of their extremely low, catalysis-incompatible rate of nucleotide–medium exchange [10]. This was the reason for their long being beyond the scope of research. Regular investigation of their properties was begun by Boyer’s team in 1987 [11]; to date, extensive experimental results in this field have been published. This review presents description, generalization, and analysis of properties of noncatalytic nucleotide binding sites and offers putative mechanisms of their involvement in nucleotide-dependent ATP synthase regulation.
ATP SYNTHASE STRUCTURE AND PRINCIPAL FUNCTIONS
Chloroplast, mitochondrial, and bacterial ATP synthases are of the FoF1-ATPase type. They have a water-soluble peripheral part (F1) and a membrane part (Fo). F1 is composed of alternating three α- and three β-subunits arranged around a double helix of the γ-subunit, plus one δ- and one ε-subunit (Fig. 1). The composition and number of subunits in the membrane part vary depending on the genesis of the organelle. The c-subunits range in number from eight (ATP synthase of animal mitochondria) to 15 (cyanobacterium Spirulina platensis) [12, 13]. Chloroplast ATP synthase incorporates a single subunit of types I, II, and IV and 14 subunits of type III [14, 15], which correspond to b-, b′-, a-, and c-subunits of bacterial ATP synthase, respectively. At the interface between α- and β-subunits there are three catalytic and three noncatalytic sites. The latter are formed mostly by amino acid residues of the α-subunit, while the former by those of the β-subunit [11, 16].
Fig. 1. Model of FoF1-ATPase.
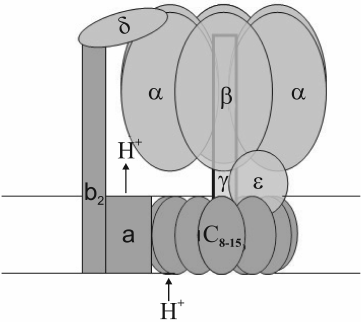
During ATP synthesis, energy transformation occurs as a two-step event. The first step consists in transmembrane proton transfer performed by the hydrophobic part (Fo) of the ATP synthase complex. Then the transmembrane difference in proton electrochemical potentials is transformed into mechanical energy of rotation of the “rotor”, i.e. a block of c-subunits associated with the γ- and ε-subunits [17, 18]. Its rotation relative to the immobile stator presented by α3β3δabb′-subunits is caused by consecutive protonation/deprotonation of a conserved carboxyl group in each c-subunit. A more detailed description of the Fo structure and the mechanism of proton transfer coupling with c-block rotation is given in [19]. At the other step, specific amino acid residues of the rotating γ-subunit consecutively interact with amino acid residues of each β-subunit, thereby inducing conformational changes of the catalytic sites; in turn, this provokes ADP- and phosphate binding followed by their conversion, to give ATP with its subsequent dissociation. Rotation of the heterohexamer α3β3 (caused by the γ-subunit rotation) is prevented by interaction of an α-subunit with the δ-subunit tightly bound to the membrane-fixed b-subunits (Fig. 1) [20]. Thus, at the last step, mechanical energy of the “rotor” is converted into chemical energy of ATP high-energy bonds. ATP hydrolysis causes reverse rotation of the “rotor”, thereby accordingly re-directing the transmembrane proton transfer.
An important prerequisite to the high catalytic activity of FoF1-ATPase is cooperative functioning of its catalytic sites that allows using the substrate binding energy for dissociation of reaction products (energy recuperation system) [21-23]. A comprehensive scheme of cooperative catalysis involving three catalytic sites and diagrams of varying energy levels of the enzyme in the course of the reaction are presented in [24-26]. As reported elsewhere [27, 28], transition from cooperative to single-site catalysis (with substrate/enzyme <1) accelerates the reaction rate by 5-6 orders of magnitude. It is generally agreed that cooperation of reaction steps occurring at the three catalytic sites is provided by the rotating γ-subunit. However, the difference between activities of F1 subcomplexes with (α3β3γ) and without (α3β3) this subunit is much less [29, 30]. This suggests involvement of α- and β-subunits whose interactions may additionally contribute to cooperative functioning of the catalytic sites. The suggestion is supported by the recently reported consecutive conformational changes of β-subunits that accompany ATP hydrolysis by γ-deficient enzyme [31].
Nucleotide-Dependent Activation/Inactivation of FoF1- and F1-ATPases
A decrease in transmembrane proton gradient induces reversible Mg2+- and ADP-dependent inactivation of FoF1-ATPases of different origin [32-35]. In the case of isolated F1-ATPase with ADP tightly bound at a catalytic site, the inactivation is initiated by Mg2+ added to the medium [36-43]. Inorganic phosphate, ATP, and some oxyanions stabilize activity of FoF1-ATPases and provide transition of inactive F1-ATPases into their active state [32-34, 36, 38-46]. Complete activation of FoF1-ATPases can be achieved at increased transmembrane proton potential. The inactivation/activation shows clear correlation with ADP tight binding/dissociation from a catalytic site [38, 47-50].
Isolated chloroplast coupling factor (CF1) used as a model for inactivation studies by pre-steady state kinetics showed that its inactivation was accompanied by differentiation of functions of its catalytic sites: one of these tightly bound MgADP, another – MgATP, while the third one lost its affinity for nucleotides [42]. This finding was supported by X-ray analysis of the mitochondrial coupling factor MF1 [51].
It is believed that the ability to tightly bind MgADP, and hence to be inactivated, is determined to a large extent by asymmetric interactions of the γ-subunit with each of the three F1 β-subunits; specifically, samples with deleted γ-subunits failed to be inactivated by MgADP [52]. On the other hand, pre-steady state kinetics of ATP hydrolysis suggested that tightness of MgADP-to-catalytic site binding, and hence the degree of inactivation, depends on ATP binding to noncatalytic sites [53, 54]. The need to find the mechanism of Mg2+- and ADP-dependent regulation of FoF1-ATPase activity dictates the demand for better knowledge of the properties of the noncatalytic sites.
STRUCTURE AND PROPERTIES OF F1-ATPase NONCATALYTIC
SITES
Structure of F1-ATPase Noncatalytic Sites
The structure of F1-ATPase noncatalytic sites bears a considerable similarity to that of catalytic sites, although it is composed mostly of amino acid residues belonging to the α-subunit. Both catalytic and noncatalytic sites are located at the interface between α- and β-subunits, and the following elements are common to them: (i) a Tyr-containing domain responsible for interaction with the adenine part of nucleotides (for noncatalytic sites of mitochondrial F1, this is βTyr368 [51]); (ii) a positively charged lysine residue (αLys175) that interacts with negatively charged oxygen atoms in the polyphosphate part of nucleotides; (iii) a threonine residue (αThr176) responsible for binding a magnesium ion that in turn interacts with the polyphosphate tail of ADP or ATP. Lys175 and Thr176 are elements of the P-loop, also called the Walker A motif (GXXXXGKT/S) that is typical for nucleotide-binding proteins [55, 56]. Through a water molecule, the magnesium ion is also bound to αAsp269 that belongs to another conserved sequence, that is, the Walker B motif [56]. Among other site-forming residues, αGln208 and βGlu188 occupy the same position at noncatalytic and catalytic sites, respectively. The latter is believed to be directly involved in catalysis as a Lewis base. Substitution of an amino group for oxygen in αGln208 and the side chain orientation opposite to that of the γ-phosphate probably explain the lack of catalytic activity shown by the noncatalytic sites. This is consistent with the fact that this activity can appear as a result of directed mutagenesis-induced substitution of glutamine acid for a glutamine residue [57].
Properties of F1-ATPase Noncatalytic Sites
In studies of properties of noncatalytic sites, the pivotal role was played by a technique developed for assessment of selective nucleotide binding to these sites. The technique is based on the significant difference between the nucleotide exchange rates at the catalytic and noncatalytic sites. The assessment is a three-step procedure; its first step is filling of all sites with radioactive nucleotides, the second is selective nucleotide replacement at the catalytic sites by non-radioactive ATP (“chase”) in the course of the ATPase reaction, and the last step is rapid (as compared to the nucleotide exchange rate at noncatalytic sites) removal of free nucleotides from the reaction mixture using forced gel filtration followed by quantitation of tightly bound nucleotides from sample radioactivity [58-61]. Another method of assessment of nucleotide incorporation into noncatalytic sites was proposed later [62]; it used tryptophan introduced by directed mutagenesis to assess its fluorescence quenching at noncatalytic sites caused by nucleotide binding.
Nucleotide–noncatalytic site interactions. Noncatalytic sites of F1-ATPases of different origin can bind as many as three molecules of ATP or two molecules of ADP per mole of the enzyme [60-62]. The nucleotide affinity for these sites strongly increases in the presence of Mg2+, which is consistent with X-ray data on incorporation of three MgATP molecules into F1 noncatalytic sites [63]. Neither free AMP nor its complex with magnesium can bind to the noncatalytic sites [62]. Apart from adenine nucleotides, GTP, GDP, and ITP are also capable of binding to the noncatalytic sites, as well as such nucleotide analogs as FSBA, AMP-PNP, 2-azido-ATP, and 2-azido-ADP [43, 51, 64-68].
It was found that not all noncatalytic sites are functionally identical. For example, only two sites of mitochondrial F1 retain nucleotides after precipitation with ammonium sulfate [58], and only two CF1 sites retain nucleotides during heat activation in the presence of ADP. In the course of enzyme incubation with ATP, the nucleotide occupies a third (vacant) site and replaces ADP at a second site [61]. The functional heterogeneity of the sites probably results from certain peculiarities of their structures. Specifically, the residue βArg372 was found solely in the site belonging to the αE-subunit (designated according to Abrahams et al. [51]), while βArg356 and βTyr368 were reported to be members of the αDP-formed site.
Unlike noncatalytic sites of mitochondrial and chloroplast F1-ATPases, the E. coli F1 sites show (according to [62, 70]) similar selectivity for ATP and ADP (for MgATP and MgADP, Kd = 20 μM). In the absence of information on the E. coli F1 spatial structure and kinetics, this difference cannot be unambiguously attributed to peculiarities of the experimental approach [71], quality of the enzyme sample used, or species specificity of F1. It is noteworthy that heterogeneity of the E. coli F1 noncatalytic sites was reported by Hyndman and colleagues who found that separately, ADP and ATP could bind to only two sites, while together, they could occupy all the three noncatalytic sites [72].
Heterogeneity of the noncatalytic sites of dithiothreitol-activated CF1 is clearly manifested in nucleotide binding kinetics. As follows from analysis of the kinetics, nucleotide binding involves both a rapid reversible stage and a slow poorly reversible one [73]. For the rapid stage (K1), equilibrium constants of two CF1 noncatalytic sites range from 1 to 9 μM, with one of these showing higher affinity for ATP and the other for ADP. The tight binding rate constants (k2) range from 0.1 to 6.7 min–1, showing an 8-fold difference between the sites for ATP binding and a 50-fold difference for ADP binding. The kinetics of nucleotide dissociation is described by the first order equation and characterized by dissociation rate constants (k3) 1·10–3 min–1 (ATP) and 1.5·10–1 min–1 (ADP) without notable difference between the sites [73]. The third noncatalytic site of CF1 was occupied by endogenous ADP and, unlike that of heat-activated enzyme [61], showed no nucleotide exchange with the medium. These data yielded the following scheme of interaction between nucleotides and CF1 noncatalytic sites:
![]()
Oxyanion–noncatalytic site interactions. Interest in interactions between F1-ATPases and oxyanions has been mostly inspired by the ability of some oxyanions (sulfite, bicarbonate, borate, phosphate, etc. [41, 44, 74, 75]) to reactivate MgADP-inactivated enzymes. The literature demonstrates that oxyanions can bind to both catalytic and noncatalytic sites. Their binding to catalytic sites is supported by competition between sulfite and phosphate during phosphorylation [76, 77], sulfite-induced inhibition of the initial stage of pre-steady state kinetics of ATP hydrolysis (without MgADP inactivation) [78], and incorporation of a sulfate or phosphate ion into the spatial structure of one of the three F1 catalytic sites [79, 80]. Oxyanion binding to noncatalytic sites is indicated by suppression of nucleotide incorporation into these sites by sulfate, sulfite, and some other hydrolysis-stimulating oxyanions [60, 81, 82]. The ability of oxyanions to bind to sites of both types raised a question as to binding to what site (or special site) causes enzyme reactivation. An analysis of the pre-steady state kinetics of Mg-dependent ATP hydrolysis by CF1-ATPase showed that the sites involved in reactivation (reaction stimulation) did not coincide with the catalytic sites [78]. Subsequent studies showed that the activation resulted from oxyanion interactions with noncatalytic sites. Specifically, it was found that oxyanion (sulfite, bicarbonate, or borate) concentrations causing half-maximal activation corresponded to those causing the half-maximal inhibition of radioactive ATP incorporation into noncatalytic sites [81]. The authors of [83] employed the ability of pyrophosphate to selectively and tightly bind to F1-ATPase noncatalytic sites [70, 84, 85]. The slow dissociation of PPi from CF1 (t1/2 = 14 min) allowed experiments aimed to reveal the effect of oxyanions using CF1 free of loosely bound PPi. Its tight binding to noncatalytic sites during preincubation suppressed the ability of sulfite and bicarbonate oxyanions to stimulate Mg-dependent ATPase activity of the enzyme, while pyrophosphate dissociation correlated with restoring of this ability, thereby demonstrating competition between PPi and these oxyanions for binding to the same site.
It was found that rate constants of ADP and ATP dissociation from noncatalytic sites of dithiothreitol-activated enzyme (0.15 and 0.001 min–1 [73]) were much lower than the rate constants of stimulation of MgADP-inactivated enzyme by oxyanions (about 1 min–1 [78]). At the same time, oxyanions appeared to be much weaker competitors than ADP and ATP [81, 86]. Since both nucleotides and oxyanions bind to noncatalytic sites, a question arises as to how oxyanions can produce a stimulating effect at high concentrations of nucleotides in the reaction mixture. As mentioned above, noncatalytic sites of CF1 and ATP synthase are heterogeneous: one of these displays high affinity for ATP, another for ADP, and a third one shows similar efficiency in binding of the two nucleotides [61]. The last two sites possess vacant sections corresponding to the position of either ATP γ-phosphate or inorganic phosphate. Because oxyanions are structurally similar to phosphate ions [87], these sections may act as oxyanion-binding sites. Since one noncatalytic site displays specificity to ADP, the second site capable of interacting with both nucleotides is a likely place of oxyanion binding. This suggestion is in agreement with the first order of the reaction of enzyme activation by sulfite [78]. However, it should be mentioned that specificity of one of the noncatalytic sites to ADP is not absolute, because long incubation with excess MgATP makes ATP incorporation into all the three sites possible [61]. Hence, at a high oxyanion concentration, its binding to the other noncatalytic site cannot be ruled out.
Role of Noncatalytic Sites in Modulation of Catalytic F1-ATPase Activity
Early evidence for noncatalytic site contribution to catalytic activity of F1 followed from the findings that covalent modification of these sites caused its inactivation [65, 67, 68] and that ATP binding to these sites influenced its GTPase activity [59]. (It was shown later that ATP-to-noncatalytic site binding is a necessary condition for manifestation of ATPase activity by CF1 and MF1 [60, 84].) Importantly, substitution of GTP for ATP even at one noncatalytic site resulted in a much lower activity level [43, 61].
It should be noted that the concept on the functional role of noncatalytic sites is not universally supported [70, 88, 89]. Specifically, it has been reported that site occupancy has no effect on “single-site” catalysis of the ATPase reaction (in conditions when only one catalytic site is occupied due to a low substrate concentration) [90]. The maximal reaction rate has been shown to be unaffected [88]. The zero effect of mutation of residue αD261N (E. coli F1 numbering) participating in magnesium binding at a noncatalytic site has been mentioned [91], as well as the absence of difference between noncatalytic site occupation with ATP or ADP during oxidative phosphorylation [91]. However, a detailed analysis of the reported data and their correlation with other research results does not rule out a functional role of the noncatalytic sites. For example, the studies of pre-steady state kinetics of MF1 ATP hydrolysis showed that the hydrolysis-stimulating effect of noncatalytic sites was observed exclusively at ATP concentrations sufficient for occupancy of these sites by ATP. This stimulation contributed to disappearance of the effect of negative cooperativity (increased Km caused by abnormal reaction acceleration at 100-300 μM ATP) [53, 92]. The absence of the effect of noncatalytic site occupancy by ADP on the reaction rate described in [88] could probably be explained by closeness of the incubation time to the rate of bound ADP/ATP exchange. Mutation of the aD261N residue might slow ATP binding to noncatalytic sites but not stop it [93]. Finally, a 2-3-fold acceleration of ATP synthesis by thermophilic bacterial ATP synthase in proteoliposomes under conditions of occupancy of noncatalytic sites with ATP has been demonstrated [94].
An analysis of pre-steady state kinetics of ATP hydrolysis by F1-ATPases showed that stimulation of the activity caused by ATP interaction with F1 noncatalytic sites resulted from a decrease in MgADP tight binding to one of the catalytic sites [53, 54]. The decreased effectiveness of this interaction attained by directed mutagenesis of one amino acid residue (D261N) at a noncatalytic site resulted in a reduced reaction rate [93], while a more profound site modification with alanine substituted for several residues (R175, T176, D261, and D262) ruled out MgADP dissociation, thereby making the inactivation virtually irreversible [92, 95, 96]. Note that the mutant was also deprived of the oxyanion-caused activation and dissociation of inhibitory MgADP [57].
The presented heterogeneity of noncatalytic sites suggested certain differences in their functional roles. However, this supposition is not wholly supported by the literature. Bullough and colleagues [68] reported that upon the interaction of FSBA with MF1, the effect of the latter correlated with modification of a third noncatalytic site, while modifications of the other two sites had no effect on either ADP-induced inactivation or negative cooperativity. A similar correlation was observed for reactivation of MgADP-inhibited MF1 caused by consecutive filling of noncatalytic sites with the nucleotides [84]. A complete inactivation of MF1-ATPase occurred after 2-azido-ATP derivatization at two noncatalytic sites [67]. Pyrophosphate-induced hydrolysis stimulation was observed after the two last noncatalytic sites had been occupied by PPi [97]. The CF1 activity was dependent on ATP inclusion in the noncatalytic site that had been vacant during heat activation of CF1 in the presence of ADP (designated as Site A). In contrast, the enzyme activity showed no dependence on the insignificant ADP-for-ATP exchange at Site C [61]. Another research team compared activity levels of the F1-ATPase α3β3γ-subcomplex from Bacillus PS3 possessing 1, 2, and 3 noncatalytic sites incapable of nucleotide binding [98]. A single functioning site was found sufficient for appearance of notable activity, while the peak was achieved with all the three sites involved. In spite of some obvious differences, these data are indicative of an effect of the state of noncatalytic sites on catalytic sites. However, evidence has been provided that properties of noncatalytic sites are dependent on the catalytic sites: during ATP hydrolysis by mitochondrial F1 and by F1 from E. coli, dissociation of the nucleotides from noncatalytic sites was accelerated 8- and 23-fold, respectively [58, 72].
PROPERTIES OF NONCATALYTIC SITES OF ATP SYNTHASE COMPLEXES
Properties of Noncatalytic Sites of Isolated ATP Synthase Complexes
Isolated ATP synthase complexes can be viewed as a transition stage between water-soluble coupling factors (that are actually model systems) and native systems including membrane-integrated complexes. In this situation, studies of the properties of noncatalytic sites within isolated ATP synthase complexes are of special interest. To date, the best-studied ATP synthase complex is that of chloroplasts [99, 100]. The reported sample contained three endogenous nucleotides: two ATPs at noncatalytic sites and one ADP at a catalytic site. By varying conditions of incubation with ADP, ATP, and its covalently binding analog 2-azido-ATP, a sample was obtained with 5 nucleotides/mole, specifically, with 3 ATPs (including the analog) at noncatalytic sites and 2 ADPs at catalytic sites. The noncatalytic sites differed much from one another as to stability of their complexes with ATP: the dissociation constant of one site was about 50 μM, that of another was about 2 μM, and the constant value for a third one was nanomolar. An interaction between the catalytic and noncatalytic sites was manifested by the fact that ADP inclusion in a catalytic site promoted ATP dissociation from a noncatalytic site.
Analysis of peptides yielded by trypsinolysis of ATP synthase complexes preincubated with covalently binding radioactive 2-azido-derivatives of ADP and ATP indicated that CFoF1 noncatalytic sites incorporate ATP but not ADP. However, this conclusion contradicts the above-described ability to bind ADP shown by noncatalytic sites of the coupling factor CF1 and by the complex of thermophilic bacteria TFoF1 [58, 70, 79, 88]. Additional evidence for the ability of CFoF1 noncatalytic sites to incorporate ADP is as follows. According to [100], up to 15-20% of 2-azido-ADP was detected at noncatalytic sites in spite of the fact that the nucleotide used for covalent modification was 2-azido-ATP. The authors of this study reported that prolonged incubation with EDTA turned tightly bound ATP into ADP. It cannot be ruled out that this amount (15-20%) of 2-azido-ADP originated from 2-azido-ATP tightly bound to the noncatalytic site. As to their previous report [99] that no 2-azido-ADP was detected at this site, it could probably be explained by a much lower stability of noncatalytic site–ADP complexes as compared to those with ATP (see above) and by ADP dissociation during free nucleotide removal by triple gel filtration using magnesium-free eluents [61, 96].
In agreement with the data on coupling factors F1 of different origin, nether 2-azido-ATP binding to noncatalytic sites nor covalent modification of the site component bTyr385 had any effect on CFoF1 activity under single-site catalysis conditions. However, in contrast to the data on F1 [53, 54, 59, 61, 98] and TFoF1 [94], occupancy of all noncatalytic sites of CFoF1 with the nucleotides had no effect on the enzymatic activity under conditions of cooperative (three sites involved) catalysis [100]. Suppression of the cooperative catalysis was observed only on condition of covalent binding of 2-azido-ATP; then, to attain complete inhibition, modification of only one site was sufficient. Accordingly, covalent modification of one site from the thermophilic bacterial ATPase complex TFoF1 resulted in complete suppression of ATP synthesis and hydrolysis in proteoliposomes [102]. It can be suggested that covalent modification of bTyr385 restricted conformational mobility of the noncatalytic site required for cooperative catalysis.
Effect of Membrane Energization on Noncatalytic Site Properties
To learn the possible role of noncatalytic sites in regulation of ATP synthase activity, it is important to know the effect of membrane energization on properties of these sites. According to [73], the complex of ATP with noncatalytic sites of isolated CF1 is quite stable (k3 = 1·10–3 min–1), which virtually rules out its involvement in regulation of this activity. As reported in [100], membrane energization by an artificial ΔpH/Δψ gradient resulted in release of a small part (about 4-6%) of 2-azido-ATP from a noncatalytic site. This was attributed to the exchange between catalytic and noncatalytic sites during ATP synthase reconstitution into liposomes and regarded as evidence that noncatalytic site properties were energization-independent. Importantly, because the membrane energization is extremely short (about 5 s), it must be taken into account if nucleotide dissociation from a noncatalytic site takes more than this time, and the result is within the limits of the experimental error.
In studies of the regulatory function of noncatalytic sites, an important step was modification of the “cold chase” technique as applied to nucleotide binding to ATP synthase of thylakoid membranes [101]. In principle, its applicability was demonstrated by the finding that in the presence of sulfite, thylakoid membranes preliminarily activated by light and dithiothreitol provide rapid ATP hydrolysis in the dark [45]. Using this technique, it was shown that in the dark the level of nucleotide incorporation into noncatalytic sites of ATP synthase of thylakoid membranes was extremely low (below 0.1 mole per mole of ATP synthase) [101]. Comparison of noncatalytic sites in terms of their exposure to nucleotide exchange with the reaction mixture revealed that this characteristic depended on closeness of CF1 to its native state. Specifically, heat-activated CF1 in the presence of ADP showed occupancy of three noncatalytic sites, in dithiothreitol-activated CF1 nucleotides occupied two sites, in isolated CFoF1 – one site, whereas CFoF1 of thylakoid membranes demonstrated their almost complete absence.
Nucleotide incorporation into noncatalytic sites can be many-fold increased by light-induced membrane energization. The observed maximal incorporation was 1.6 nmol/mg of chlorophyll, which, taking into account the reported value for CFoF1 (1 nmol/mg of chlorophyll) [45, 103] and possible losses [104], corresponds to a value of 2 moles per mole of enzyme. The incorporated nucleotides were both ADP and ATP [105]. A third noncatalytic site of ATP synthase was probably uninvolved in the energy-dependent exchange. This supposition agrees with high stability of the noncatalytic site–ATP complex reported for isolated CFoF1 [100] and with the fact that the ATP synthase complex of thylakoid membranes retained tightly bound ATP after light-induced activation in the presence of thioredoxin and dithiothreitol followed by free nucleotide removal [106].
Interestingly, a rather high level of dark inclusion of nucleotides into CFoF1 was attained on condition of prior nucleotide removal from noncatalytic sites [107]. Hence, the pivotal step of their interaction with ATP synthase noncatalytic sites is dissociation of the bound nucleotide. The rate of energy-dependent dissociation notably decreased with increasing Mg2+ concentration. With 1 mM MgCl2, the dissociation rate constants of ADP and ATP were close to each other and amounted to 5.7 and 5.0 min–1, respectively [105]. Of note, these values are almost two orders of magnitude lower than the rate constants of energy-dependent dissociation of ADP tightly bound at a catalytic site [50].
Heterogeneity and interplay of noncatalytic sites described above for coupling factors and ATP synthase complexes of different genesis distinctly manifested themselves under conditions of membrane energization. At varying nucleotide concentrations in the reaction mixture and a high ATP/ADP ratio, the amount of ATP incorporated into noncatalytic sites depended on the amount of bound ADP. With excess ADP, this dependence was not observed [107]. The increasing ATP/ADP ratio with their unchanged total amount resulted in a reduced total incorporation of the nucleotides into ATP synthase [105]. This suggests cooperative functioning of two noncatalytic sites. ADP binding at one of them (Site B or 5, as designated by [61] or [100], respectively) is a prerequisite to filling the other site (Site A or 6) with ATP. However, ADP binding to Site 6 does not require ATP incorporation into Site 5. At similar concentrations of ATP and ADP in the reaction mixture, the dissociation constant of ATP-to-Site 6 binding is similar to that of ADP-to-Site 5 binding (2.0 ± 0.3 μM). With excess ATP, its binding to Site 6 is characterized by Kd = 33 ± 8 μM, while this parameter for ADP binding to this site with an excess of ADP is Kd = 38 ± 18 μM [108].
The kinetic characteristics of energy-dependent nucleotide exchange at noncatalytic sites are quite close to those of light-induced ATPase activation/inactivation of chloroplast ATP synthase. Indeed, the constant of ADP dissociation from Site 6 (38 ± 18 μM) corresponds (within experimental error) to that of the site responsible for suppression of ATPase activity caused by light preincubation in the presence of ADP (45 ± 12 μM) [108]. The rate constant of inactivation (1 ± 0.2 min–1) agrees with the time of half-maximal exchange at noncatalytic sites (about 1 min) [107], which, in turn, is equal to the time of light preincubation with ATP required for ATPase activity stimulation [109]. Lastly, the constant of ATP dissociation from the stimulation-responsible site (27 ± 2 μM) [108] is close to that of ATP dissociation from Site 6 (33 ± 8 μM). The mentioned correlations are evidence for presumable responsibility of this noncatalytic site (Site 6) for regulation of the ATPase activity of ATP synthase.
With the above taken into account, the following mechanism of reversible inactivation of F1- and FoF1-ATPase can be proposed (Fig. 2).
Fig. 2. Scheme of inactivation/activation of chloroplast CF1-ATPase. NS and CS, noncatalytic and catalytic sites, respectively; ADPtb, tightly bound ADP. Empty box denotes either a vacant catalytic site or a site with loosely bound ATP. The proportion of these states is ATP concentration-dependent.
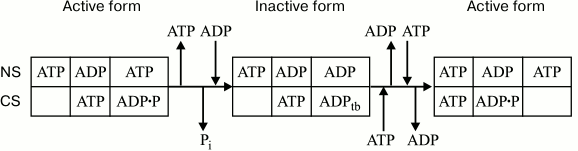
The active state of the enzyme implies ATP bound to a noncatalytic site that is capable of interacting with both nucleotides. Under membrane energization conditions, the exchange of ADP for ATP causes a higher ADP affinity for a catalytic site and its tight binding, which results in enzyme inactivation. With excess ATP, membrane energization accelerates the ADP–ATP exchange at noncatalytic sites, thereby causing enzyme reactivation.
MECHANISMS OF INVOLVEMENT OF NONCATALYTIC SITES IN REGULATION OF
FoF1-ATPase ACTIVITY
Dependence of catalytic activity of FoF1-ATPases on the nature of noncatalytic site-bound nucleotides implies the presence of molecular structures responsible for conformational signal transduction between noncatalytic and catalytic sites. Search for these structures was based on the ability of noncatalytic sites to influence the cooperative properties of the enzyme without affecting single-site catalysis [54, 92]. Logically, these structures were supposed to comprise amino acid residues inherent to sites of both types, because even slight changes in residue coordinates caused notable activity changes. It was suggested [110] that such a structure could be represented by the short sequence βT354-βY345 between residue αQ172 (via H-bonding with βT354) from a noncatalytic site and residue βY345 from a catalytic site (mitochondrial F1 numbering). Additionally, this link is supported by the αR171–βD352 salt bridge. Mutation of residue αQ172 from the Walker A motif was observed to suppress a negative cooperativity effect in ATP binding.
Another hypothesis [111] on signal transduction between noncatalytic and catalytic sites is underlain by the notion that separate parts of a protein molecule possess special degrees of freedom and can perform transduction of the conformational signal without energy losses from one part of the molecule to another (by analogy with a coated wire rope) [112]. Minimal energy dissipation occurs on condition of a minimal linker length. This requirement is met by inner sequences whose length is close to the minimal distance between a catalytic and a noncatalytic site. According to [51], for mitochondrial F1 this distance is 27 Å. Hence, with an average amino acid length of 3.6 Å [113], a sequence that links neighboring nucleotide binding sites must include at least eight amino acid residues, or more, with protein packing density taken into account. One of such sequences is the β-subunit segment between Y345 and R356, which is quite similar to the sequence described in [110] (Fig. 3).
Fig. 3. Amino acid sequences linking a catalytic site (CS) with a noncatalytic site (NS). Conserved residues are shown in black. (Adapted from [111]).

The virtually perfect identity of β-subunit sequences of different genesis within this segment is indicative of its conserved nature (Fig. 3). It meets the requirement of energy conservation during impulse transduction from its one end to the other [112] because all its residues, except D352, are not involved in Coulomb interactions with neighboring residues, and D352 through its interaction with R171 can pass a signal to Q172 that shares with R356 a position at the phosphate end of a nucleotide within the same noncatalytic site. Residue Y345 is the member of a catalytic site [51], where its position is closer than 3.5 Å from the purine part of adenine nucleotide (Fig. 4a). Residue R356 belongs to a noncatalytic site and is located at a similar distance from ATP γ-phosphate. Supposedly, Y345 provides hydrophobic environment of adenine [70, 114]; the energy of its interaction with ATP is 1.5 kcal/mol [70]. Environmental hydrophobicity increased by phenylalanine substitution for tyrosine causes a decrease in MgADP-dependent inactivation of the enzyme [114]. Still lower inactivation was observed as a result of replacement of ATP by ε-ATP showing higher hydrophobicity [115]. Evaluation of the Y345 replacement effect on catalytic activity revealed a role of the residue size (affinity decrease in the order of Tyr > Leu > Ala was more than 10-fold) [116]. The effect of R356 replacement by other residues has not been examined, although it is worth noting that its homolog αR373 similarly located at a catalytic site showed elevated mobility (a shift by 1.5 Å) upon ATP–ADP exchange [70].
Fig. 4. Interaction between catalytic and noncatalytic sites in F1-ATPase. a) Amino acid sequence βTyr345-βArg356 (atoms of terminal amino acid residues are shown as spheres). b) Amino acid sequences αSer344-αPhe357, αPro363-αArg373, and βTyr345-βArg356. The dashed line represents Coulomb interactions and H-bonding. c) Scheme of interactions between F1 catalytic and noncatalytic sites (see Fig. 1, view from above). Segments belonging to the β-subunits are shown as solid lines, and those of the α-subunits are represented by dashed lines. The subunits are designated according to [51]. (Adapted from [111]).
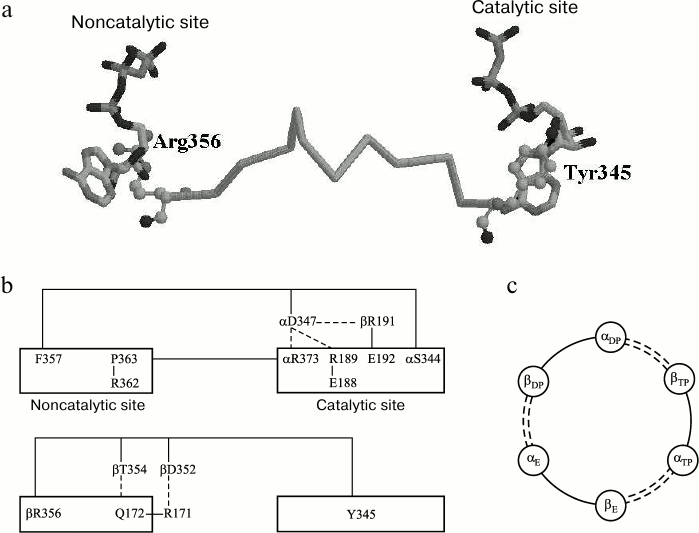
In the context of the proposed mechanism, the stimulating activity of ATP and oxyanions can be explained as follows. Upon ATP binding to a noncatalytic site, the appearance of γ-phosphate can result in a changed position of R356. This motion is imparted via S355-P346 to Y345. The protein packing density prevents cross deformations of the chain accompanied by energy loss. The position of Y345 changes with changing hydrophobic interactions with the purine part of ADP tightly bound at a catalytic site; in turn, the lower binding level causes enzyme transition from its inactive to active state. A similar effect can be produced by oxyanions that, according to [81, 83], replace the γ-phosphate at a noncatalytic site.
Two other segments linking a noncatalytic site to the neighboring catalytic site belong to the α-subunit. As seen from Fig. 3, these are conserved too. One of them begins with F357 near the ATP ribose ring at the noncatalytic site and ends with S344 close to the ATP γ-phosphate of the catalytic site (Fig. 4b). Substitution of cysteine for αF357 and its adjacent βR372 lessens dissociation ability of MgADP previously induced by ATP binding to the noncatalytic site, whereas disulfide bond-dependent mobility limitation stops the dissociation completely [117]. The αS344F mutation decreases catalytic activity of the enzyme many-fold [118]. A member of this segment, αD347, is tightly bound to αR373, βR189, and βE192 that belong to the catalytic site. Residue αR373 is the terminal member of another segment that links the catalytic site with P363 located at the noncatalytic site near ATP adenine. According to [119], R373 is not immediately involved in the catalytic cycle but responsible for cooperative functioning of the three catalytic sites. The mentioned segments also incorporate several amino acid residues the effect of which on properties of bacterial F1 has been studied using the point mutations technique, namely, αS347F, αG351D and αS373F, and αS375F (E. coli F1 numbering) [67, 120, 121]. In mitochondrial F1, their corresponding residues are S344 and G348 from one α-subunit segment and S370 and S372 from the other (Fig. 3). It is of interest that in spite of different positions and dissimilar nature of the residues, their replacements gave similar results. All these mutations, like R373 replacement, resulted in a changed catalytic cooperativity with a minor effect on nucleotide binding [117, 118]. The common feature of all these mutations was that the substituted residue was much smaller in size. The reported effects are in good agreement with the concept of conformational signal transduction that implies an increased difficulty in movement of the polypeptide chain segment with an enlarged residue, and hence, an altered position of its terminal residue relative to the closest nucleotide section.
As seen from Fig. 4c, the described segments form a system of links between all catalytic and noncatalytic sites. The proposed scheme explains recent results on the possibility of cooperative, continuous functioning of F1-ATPase catalytic sites either with a γ-subunit truncated from both its termini or without it [31, 122-124].
Regardless of abundant information available at present about properties of noncatalytic sites, some problems concerning their functional mechanisms are still to be resolved. The structural changes causing their different exposure upon transition from an isolated enzyme to the membrane-integrated ATP synthase complex are so far obscure. The mechanism of energy-dependent acceleration of nucleotide exchange at noncatalytic sites is not known either. The hypothesis on existence and functions of short conserved amino acid sequences linking catalytic and noncatalytic sites requires further support. For a long time, the extremely low rate of nucleotide exchange at noncatalytic sites was thought to either eliminate them from possession of any function or presupposed only a structural role [70]. However, preservation of these sites in the course of prolonged evolution propelled more thorough studies of their properties. The fact of many-fold acceleration of the nucleotide exchange upon energization of chloroplast thylakoid membranes showed the probability of their involvement in regulation of ATP synthase ATPase activity. Although unsupported experimentally for mitochondrial and bacterial membranes (mostly owing to the lack of appropriate techniques), the high similarity of the properties of F1-ATPase noncatalytic sites suggests their analogous behavior irrespective of genesis. Experiments on thylakoid membranes showed that the role of noncatalytic sites consists in regulation of the efficiency of MgADP-dependent inactivation, because this inactivation reaches its maximum upon ADP binding at noncatalytic sites and damps sharply upon the ATP–ADP exchange.
It is believed that the physiological sense of regulation of ATP synthase activity consists in prevention of unproductive ATP hydrolysis in conditions of an insufficient transmembrane proton gradient. However, even in the case of virtually complete inactivation, there is a minor activity level, which varies in accordance with the medium redox state, the ADP/ATP ratio, and the presence of endogenous stimulators of ATP hydrolysis. Presumably, the physiological importance of regulation of the ATP synthase activity within this range consists in maintaining a certain minor transmembrane potential required for transport of ions, polypeptides, and/or metabolites in the situation of insufficient supply of energy substrates (NADH, succinate, light).
REFERENCES
1.Stocker, A., Keis, S., Vonck, J., and Cook, G. M.
(2007) Structure, 15, 904-914.
2.Rees, D. M., Leslie, A. G. W., and Walker, J. E.
(2009) Proc. Natl. Acad. Sci. USA, 106, 21597-21601.
3.Dautant, A., Velours, J., and Giraud, M. F. (2010)
J. Biol. Chem., 285, 29502-29510.
4.Giraud, M. F., Paumard, P., Sanchez, C., Brethes,
D., Velours, J., and Dautant, A. (2012) J. Struct. Biol.,
177, 490-497.
5.Groth, G., and Pohl, E. (2001) J. Biol.
Chem., 276, 1345-1352.
6.Iino, R., Hasegawa, R., Tabata, K. V., and Noji, H.
(2009) J. Biol. Chem., 284, 17457-17464.
7.Kadoya, F., Kato, S., Watanabe, K., and
Kato-Yamada, Y. (2011) Biochem. J., 437, 135-140.
8.Corvest, V., Sigalat, C., and Haraux, F.
(2007) Biochemistry, 46, 8680-8688.
9.Konno, H., Murakami-Fuse, T., Fujii, F., Koyama,
F., Ueoka-Nakanishi, H., Pack, C. G., Kinjo, M., and Hisabori, T.
(2006) EMBO J., 25, 4596-4604.
10.Kironde, F. A. S., and Cross, R. L. (1987) J.
Biol. Chem., 262, 3488-3495.
11.Xue, Z., Zhou, J.-M., Melese, T., Cross, R. L.,
and Boyer, P. D. (1987) Biochemistry, 26,
3749-3753.
12.Watt, I. N., Montgomery, M. G., Runswick, M. J.,
Leslie, A. G., and Walker, J. E. (2010) Proc. Natl. Acad. Sci.
USA, 107, 16823-16827.
13.Pogoryelov, D., Yildiz, O., Faraldo-Gomez, J. D.,
and Meier, T. (2009) Nat. Struct. Mol. Biol., 16,
1068-1073.
14.Seelert, H., Poetsch, A., Dencher, N. A., Engel,
A., Stahlberg, H., and Mueller, D. J. (2000) Nature, 405,
418-419.
15.Varco-Merth, B., Fromme, R., Wang, M., and
Fromme, P. (2008) Biochim. Biophys. Acta,
1777, 605-612.
16.Wise, J. G., Hicke, B. J., and Boyer, P. D.
(1987) FEBS Lett., 223, 395-401.
17.Boyer, P. D. (1993) Biochim. Biophys.
Acta, 1140, 215-250.
18.Senior, A. E., Nadanaciva, S., and Weber, J.
(2002) Biochim. Biophys. Acta, 1553, 188-211.
19.Weber, J., and Senior, A. E. (2003) FEBS
Lett., 545, 61-70.
20.Weber, J., Wilke-Mounts, S., and Senior, A. E.
(2002) J. Biol. Chem., 277, 18390-18396.
21.Repke, K. R. H., and Schon, R. (1974) Acta.
Biol. Med. Germ., 33, K27-K38.
22.Blumenfeld, L. A. (1977) Problems of
Biological Physics [in Russian], Nauka, Moscow.
23.Kayalar, C., Rosing, J., and Boyer, P. D. (1977)
J. Biol. Chem., 252, 2486-2491.
24.Boyer, P. D. (2002) J. Biol. Chem.,
277, 39045-39061.
25.Futai, M., Nakanishi-Matsui, M., Okamoto, H.,
Sekiya, M., and Nakamoto, R. K. (2012) Biochim. Biophys.
Acta, 1817, 1711-1721.
26.Tikhonov, A. N., Pogrebnaya, A. F., and
Romanovsky, Y. M. (2003) Biophysics, 48, 1052-1070.
27.Grubmeyer, C., and Penefsky, H. S. (1981) J.
Biol. Chem., 256, 3728-3734.
28.Duncan, T. M., and Senior, A. E. (1985)
J. Biol. Chem., 260, 4901-4907.
29.Gromet-Elhanan, Z., and Avital, S. (1992)
Biochim. Biophys. Acta, 1102, 379-385.
30.Gao, F., Lipscomb, B., and Richter, M. L.
(1995) J. Biol. Chem., 270, 9763-9769.
31.Iino, R., and Noji, H. (2012) Biochim.
Biophys. Acta, 1817, 1732-1739.
32.Carmeli, C., and Lifshitz, Y. (1972) Biochim.
Biophys. Acta, 267, 86-95.
33.Bakker-Gruenvald, T., and Van Dam, K.
(1974) Biochim. Biophys. Acta, 347, 290-298.
34.Fitin, A., Vasilyeva, E. A., and Vinogradov, A.
D. (1979) Biochem. Biophys. Res. Commun.,
86, 434-439.
35.Vinogradov, A. D., and Zharova, T. V. (2004)
J. Biol. Chem., 279, 12319-12324.
36.Moyle, J., and Mitchell, P. (1975) FEBS
Lett., 56, 55-61.
37.Malyan, A. N., and Makarov, A. D. (1976)
Biokhimiya, 41, 888-893.
38.Minkov, I. B., Vasilyeva, E. A., Fitin, A. F.,
and Vinogradov, A. D. (1979) Biochem. Biophys. Res. Commun.,
89, 1300-1306.
39.Malyan, A. N. (1981) Photosynthetica,
15, 474-483.
40.Malyan, A. N., and Vitseva, O. I. (1983)
Biokhimiya, 48, 618-623.
41.Ebel, R. E., and Lardy, H. A. (1975) J.
Biol. Chem., 250, 191-196.
42.Malyan, A. N., and Vitseva, O. I. (1990)
Photosynthetica, 24, 613-622.
43.Guerrero, K. J., Xue, Z., and Boyer, P. (1990)
J. Biol. Chem., 265, 16280-16287.
44.Malyan, A. N., and Akulova, E. A. (1978)
Biokhimiya, 43, 952-956.
45.Larson, E. M., and Jagendorf, A. T. (1989)
Biochim. Biophys. Acta, 973, 67-77.
46.Cappellini, P., Turina, P. Fregni, V., and
Melandri, A. (1997) Eur. J. Biochem., 248, 496-506.
47.Strotmann, H., and Bickel-Sandkoetter, S. (1977)
Biochim. Biophys. Acta, 460, 126-135.
48.Bar-Zwi D., and Shavit, N. (1982) Biochim.
Biophys. Acta, 681, 451-458.
49.Drobinskaya, I. E., Kozlov, I. A., Murataliev, M.
B., and Vulfson, E. N. (1985) FEBS Lett., 182,
419-423.
50.Lohse, D., and Strotmann, H. (1989) Biochim.
Biophys. Acta, 976, 94-101.
51.Abrahams, J. P., Leslie, A. G. W., Lutter, R.,
and Walker, J. E. (1994) Nature, 370, 621-628.
52.Sokolov, M., and Gromet-Elhanan, Z. (1996)
Biochemistry, 35, 1242-1248.
53.Murataliev, M. B., and Boyer, P. D. (1992)
Eur. J. Biochem., 209, 681-687.
54.Jault, J. M., and Allison, W. S. (1993) J.
Biol. Chem., 268, 1558-1566.
55.Saraste, M., Sibbald, P. R., and Wittinghoffer,
A. (1990) Trends Biochem. Sci., 15, 430-434.
56.Walker, J. E., Saraste, M., Runswick, M. J., and
Gay, N. J. (1982) EMBO J., 1, 945-951.
57.Matsui, T., Jault, J.-M., Allison, W. S., and
Yoshida, M. (1996) Biochem. Biophys. Res. Commun.,
220, 94-97.
58.Penefsky, Y. S. (1977) J. Biol. Chem.,
252, 2891-2899.
59.Xue, Z., and Boyer, P. D. (1989) Eur. J.
Biochem., 179, 677-681.
60.Milgrom, Y. M., Ehler, L. L., and Boyer, P. D.
(1990) J. Biol. Chem., 265, 18725-18728.
61.Milgrom, Y. M., Ehler, L. L., and Boyer, P. D.
(1991) J. Biol. Chem., 266, 11551-11558.
62.Weber, J., Wilke-Mounts, S., Grell, E., and
Senior, A. E. (1994) J. Biol. Chem., 269,
11261-11268.
63.Bowler, M. W., Montgomery, M. G., Leslie, A. G.
W., and Walker, J. (2007) J. Biol. Chem., 282,
14238-14242.
64.Abbott, M. S., Czarnecki, J. J., and Selman, B.
R. (1984) J. Biol. Chem., 259, 12271-12278.
65.Bullough, D. A., and Allison, W. S. (1986) J.
Biol. Chem., 261, 5722-5730.
66.Xue, Z., Miller, C. G., Zhou, J.-M., and Boyer,
P. D. (1987) FEBS Lett., 223, 391-394.
67.Cross, R. L., Cunningham, D., Miller, C. G., Xue, Z., Zhou,
J.-M., and Boyer, P. D. (1987) Proc. Natl. Acad. Sci. USA,
84, 5715-5719.
68.Bullough, D. A., Brown, E. L., Saario, J. D., and
Allison, W. S. (1988) J. Biol. Chem., 263,
14053-14060.
69.Weber, J., and Senior, A. E. (1995) J. Biol.
Chem., 270, 12653-12658.
70.Weber, J., and Senior, A. E. (1997) Biochim.
Biophys. Acta, 1319, 19-58.
71.Bulygin, V. V., and Milgrom, Y. M. (2007)
Proc. Natl. Acad. Sci. USA, 104, 4327-4331.
72.Hyndman, D. J., Milgrom, Y. M., Bramhall, E. A.,
and Cross, R. L. (1994) J. Biol. Chem.,
269, 28871-28877.
73.Malyan, A. N., and Allison, W. S. (2002)
Biochim. Biophys. Acta, 1554, 153-158.
74.Nelson, N., Nelson, H., and Racker, E. (1972)
J. Biol. Chem., 247, 6606-6510.
75.Mitchell, P., and Moyle, J. (1971)
Bioenergetics, 2, 1-11.
76.Malyan, A. N., Kuzmin, A. N., and Vitseva, O. I.
(1990) Photosynthetica, 24, 613-622.
77.Cappelini, P., Turina, P., Fregni, V., and
Melandri, B. A. (1997) Eur. J. Biochem., 248,
496-506.
78.Malyan, A. N., and Vitseva, O. I. (1987)
Fiziol. Biokhim. Kult. Rast., 19, 456-461.
79.Menz, R. I., Walker, J. E., and Leslie, A. G.
(2001) Cell, 106, 331-341.
80.Kabaleeswaran, V., Puri, N., Walker, J. E.,
Leslie, A. G., and Mueller, D. M. (2006) EMBO J.,
25, 5433-5442.
81.Malyan, A. N. (2003) Biochim. Biophys.
Acta, 1607, 161-166.
82.Malyan, A. N., and Vitseva, O. I. (2001)
Biochemistry (Moscow), 66, 410-414.
83.Malyan, A. N. (2013) Dokl. Biochem.
Biophys., 450, 123-125.
84.Milgrom, Y. M., and Cross, R. L. (1993) J.
Biol. Chem., 268, 23179-23185.
85.Murataliev, M. B. (1992) Biochemistry,
31, 12885-12892.
86.Pronin, A. S., and Malyan, A. N. (2009)
Biochemistry (Moscow), 74, 775-780.
87.Rectenwald, D., and Hess, D. O. (1977) FEBS
Lett., 76, 25-28.
88.Wise, J. G., and Senior, A. E. (1985)
Biochemistry, 24, 6949-6954.
89.Bulygin, V. V., and Milgrom, Y. M. (2010)
Biochemistry (Moscow), 75, 327-335.
90.Senior, A. E., Lee, R. S. F., Al-Shavi, M. K.,
and Weber, J. (1992) Arch. Biochem. Biophys., 297,
340-344.
91.Weber, J., Bowman, S., Wilke-Mounts, S., and
Senior, A. E. (1995) J. Biol. Chem., 270,
21045-21049.
92.Ono, S., Hara, K. Y., Hirao, J., Matsui, T.,
Noji, H., Yoshida, M., and Muneyuki, E. (2003) Biochim.
Biophys. Acta, 1607, 35-44.
93.Jault, J.-M., Matsui, T., Jault, F. M., Kaibara, C., Muneyuki,
E., Yoshida, M., Kagawa, Y., and Allison, W. S. (1995) Proc. Natl.
Acad. Sci. USA, 34, 16412-16418.
94.Richard, P., Pitard, B., and Rigaud, J.-L.
(1995) J. Biol. Chem., 270, 21571-21578.
95.Matsui, T., Muneyuki, E., Honda, M., Allison, W.
S., Dou, C., and Yoshida, M. (1997) J. Biol.
Chem., 272, 8215-8221.
96.Dou, C., Grodsky, N. B., Matsui, T., Yoshida, M.,
and Allison, W. S. (1997) Biochemistry,
36, 3719-3727.
97.Jault, J. M., Paik, S. R., Grodsky, N. B., and
Allison, W. S. (1994) Biochemistry, 33, 14979-14985.
98.Amano, T., Matsui, T., Muneyuki, E., Hara, K., and Yoshida, M.
(1999) Biochem. J., 343, 135-138.
99.Possmayer, F. E., Hartog, A. F., Berden, J. A.,
and Graeber, P. (2000) Biochim. Biophys. Acta,
1456, 77-98.
100.Possmayer, F. E., Hartog, A. F., Berden, J. A.,
and Graeber, P. (2001) Biochim. Biophys. Acta, 1510,
378-400.
101.Malyan, A. N. (2005) Biochemistry
(Moscow), 70, 1245-1250.
102.Richard, P. (1996) Biochim. Biophys. Acta,
1275, 141-144.
103.Du, Z., and Boyer, P. D. (1990)
Biochemistry, 29, 402-407.
104.Harris, D. A., and Slater, E. C. (1975)
Biochim. Biophys. Acta, 387, 335-348.
105.Malyan, A. N. (2006) Photosynth.
Res., 88, 9-18.
106.Labahn, A., and Graeber, P. (1993) Biochim.
Biophys. Acta, 1144, 170-176.
107.Malyan, A. N. (2007) Biochemistry
(Moscow), 72, 728-734.
108.Malyan, A. N. (2010) Photosynth.
Res., 105, 243-248.
109.Shigalova, T., Lehmann, U., Krevet, M., and Strotmann, H.
(1985) Biochim. Biophys. Acta, 809, 57-65.
110.Falson, P., Goffeau, F., Boutry, M., and
Jaoult, J. M. (2004) Biochim. Biophys. Acta, 1658,
133-140.
111.Malyan, A. N. (2010) Biochemistry
(Moscow), 75, 81-84.
112.Chernavsky, D. S., and Chernavskaya, N. M.
(1999) Protein Machine. Biological Macromolecular Constructions
[in Russian], Moscow State University, Moscow.
113.Lehninger, A. L. (1972) Biochemistry,
Worth Publishers Inc., New York.
114.Wise, J. G. (1990) J. Biol. Chem.,
265, 10403-10409.
115.Schaefer, H.-J., Mueller, H. W., and Dose,
K. (1980) Biochem. Biophys. Res. Commun., 95,
1113-1118.
116.Weber, J., Lee, R. S., Grell, E., and Senior,
A. E. (1992) J. Biol. Chem., 267,
1712-1718.
117.Bandyopadhyay, S., Ren, H., Wang, C. S., and
Allison, W. S. (2002) Biochemistry, 41,
3226-3234.
118.Maggio, M. B., Pagan, J., Personage, D., Hatch,
L., and Senior, A. (1987) J. Biol. Chem.,
262, 8981-8984.
119.Le, N. P., Omote, H., Wada, Y., Al-Shawi, M.
K., Nakamoto, R. K., and Futai, M. (2000) Biochemistry,
39, 2778-2783.
120.Kanazawa, H., Noumi, T., Matsuoka, I., Hirata,
T., and Futai, M. (1984) Arch. Biochem. Biophys., 228,
258-269.
121.Wise, J. G., Latchney, L. R., Ferguson, A. M.,
and Senior, A. E. (1984) Biochemistry, 23, 1426-1432.
122.Mnatsakanyan, N., Hook, J. A., Quisenberry, L.,
and Weber, J. (2009) J. Biol. Chem., 284,
26519-26525.
123.Kohori, A., Chiwata, R., Hossain, M. D.,
Furuike, S., Shiroguchi, K., Adachi, K., Yoshida, M., and
Kinosita, K., Jr. (2011) Biophys. J., 101,
188-195.
124.Usukura, E., Suzuki, T., Furuike, S., Soga, N.,
Saita, E. I., Hisabori, T., Kinosita, K., Jr., and Yoshida, M. J.
(2012) J. Biol. Chem., 287, 1884-1891.