Mechanism of Saccharomyces cerevisiae Yeast Cell Death Induced by Heat Shock. Effect of Cycloheximide on Thermotolerance
E. G. Rikhvanov, I. V. Fedoseeva*, N. N. Varakina, T. M. Rusaleva, and A. V. Fedyaeva
Siberian Institute of Plant Physiology and Biochemistry, Siberian Division of the Russian Academy of Sciences, ul. Lermontova 132, 664033 Irkutsk, Russia; fax: (3952) 517-054; E-mail: fedoseeva@sifibr.irk.ru; eugene@sifibr.irk.ru; fedyaeva.anna@mail.ru* To whom correspondence should be addressed.
Received August 8, 2013; Revision received September 24, 2013
The mechanism of yeast cell death induced by heat shock was found to be dependent on the intensity of heat exposure. Moderate (45°C) heat shock strongly increased the generation of reactive oxygen species (ROS) and cell death. Pretreatment with cycloheximide (at 30°C) suppressed cell death, but produced no effect on ROS production. The protective effect was absent if cycloheximide was added immediately before heat exposure and the cells were incubated with the drug during the heat treatment and recovery period. The rate of ROS production and protective effect of cycloheximide on viability were significantly decreased in the case of severe (50°C) heat shock. Treatment with cycloheximide at 39°C inhibited the induction of Hsp104 synthesis and suppressed the development of induced thermotolerance to severe shock (50°C), but it had no effect on induced thermotolerance to moderate (45°C) heat shock. At the same time, Hsp104 effectively protected cells from death independently of the intensity of heat exposure. These data indicate that moderate heat shock induced programmed cell death in the yeast cells, and cycloheximide suppressed this process by inhibiting general synthesis of proteins.
KEY WORDS: cycloheximide, Saccharomyces cerevisiae, thermotolerance, Hsp104, reactive oxygen species, programmed cell deathDOI: 10.1134/S0006297914010039
Abbreviations: CFU, colony-forming units; DCF, 2′,7′-dichlorofluorescein; Dnm1, dynamin 1; H2DCF·DA, 2′,7′-dichlorodihydrofluorescein diacetate; Hsps, heat shock proteins; PCD, programmed cell death; ROS, reactive oxygen species; SDS, sodium dodecyl sulfate; Yca1 (MCA1), yeast caspase 1.
The ability of yeast cells to withstand the damaging effect of
short-term exposure to high temperature is determined by basal
thermotolerance. If cells are subjected to mild heat treatment at
37-39°C (heat stress), synthesis of heat-shock proteins (Hsps) is
induced, and ability of the cells to withstand the damaging effects of
high temperature (heat shock) is increased. This phenomenon is known as
induced or acquired thermotolerance [1, 2]. The relationship between the appearance of Hsps
and thermotolerance motivated the search for the causal connection
between these two phenomena. According to [3], the
suppression of Hsp synthesis by the protein synthesis inhibitor
cycloheximide reduced the development of induced thermotolerance in
Saccharomyces cerevisiae cells. Inhibition of induced
thermotolerance by cycloheximide has been confirmed by other authors
using the same biological system [4, 5]. These facts indicate that de novo protein
synthesis is required for induced thermotolerance. However, the
relationship between Hsp synthesis and induced thermotolerance was not
always straightforward. Other researchers failed to observe the
negative effect of cycloheximide on induced thermotolerance in yeast
cells [6, 7]. Contradictory
effects of cycloheximide cast doubt on the notion that Hsps have
protective function. Some authors had supposed that Hsps are merely
“cell knee-jerk reflex on stimulus” [1]. The generation of S. cerevisiae mutants
that do not synthesize Hsps clarified the role of Hsps in induced
thermotolerance. The loss of HSP104 [2] or
suppression of Hsp104 synthesis [8] was shown to
impair the ability of S. cerevisiae cells to withstand lethal
heat shock. Hsp104 promotes disaggregation and refolding of damaged
proteins, the increase in its content being one of the main factors
determining induced thermotolerance in yeast cells [2, 9]. However, the reasons for
the contradictory data concerning the effect of cycloheximide on
induced thermotolerance remained unknown.
Heat shock can induce the development of programmed cell death (PCD) in mammalian cells [10]. PCD is a specific suicide program that is characterized by externalization of phosphatidylserine to the outer leaflet of the plasma membrane, chromatin condensation, DNA fragmentation, increased generation of reactive oxygen species (ROS), fragmentation of mitochondria, and release of cytochrome c from mitochondria into the cytosol [11]. Several pieces of indirect evidence indicate that heat shock also induces PCD in S. cerevisiae cells. Yeast cells expressing the human anti-apoptotic protein Bcl-2 retained viability under conditions of otherwise lethal heat shock [12]. A number of proteins involved in the development of PCD were found in the yeast cells. The loss of these proteins inhibits the process. PCD in yeast implicates Yca1 – a Ca2+-dependent cysteine protease; Dnm1 – a homolog of an animal protein (Drp1, dynamin related protein) responsible for the fragmentation of mitochondria; Nma111 (nuclear mediator of apoptosis) – an ortholog of animal mitochondrial serine proteases; Ndi1 (NADH dehydrogenase internal) – an internal NADH dehydrogenase; Mmi1 (microtubule and mitochondria interacting protein) – a homolog of human protein TCTP (translationally controlled tumor protein); Tat-D (twin arginine translocation D, YBL055C) – a nuclease; Stm1 (suppressor of Tom1) – a protein involved in translation. At the same time, the loss of these proteins was found to protect yeast cells from death induced by heat shock [13, 14]. On the other hand, the loss of Bxi1, a homolog of mammalian anti-apoptotic protein Bi-1 (Bax inhibitor-1), reduced thermotolerance of yeast cells and increased their susceptibility to ethanol, a known trigger of PCD [15]. However, it remains unknown whether or not heat shock induces PCD in yeast cells.
The ability of cycloheximide to inhibit PCD is a well-described physiological effect of the drug. Treatment with cycloheximide was shown to inhibit PCD in yeast cells induced by low concentrations of hydrogen peroxide [16], acetic acid [17], and α-factor [18]. Higher concentrations of hydrogen peroxide and acetic acid resulted in necrosis, which was not inhibited by cycloheximide [16, 17]. Therefore, prevention of cell death as a result of cycloheximide treatment is considered as a specific indicator differentiating between PCD and necrosis [16-18].
Based on these facts, it is logical to assume that in yeast as well as in mammalian cells [10] cell death can occur via PCD or necrosis depending on the intensity of heat exposure. Therefore, PCD should be antagonized by cycloheximide treatment, while necrosis should not. The authors studying the effect of cycloheximide on induced thermotolerance did not take into account the possibility that cell death could occur via the PCD or necrosis pathway. Therefore, the goal of this work was to study the effect of cycloheximide on basal and induced thermotolerance in S. cerevisiae cells depending on intensity of the heat exposure.
MATERIALS AND METHODS
Yeast strains and growth conditions. Parent type S. cerevisiae strain Ψ-74-D694 (MATa, ade1-14(UGA), trp1-289(UAG), his3Δ-200, ura3-52, leu2-3, 112 [psi–]) and its isogenic mutant Ψ-74-D694::hspΔ-1L (MATa, ade1-14(UGA), trp1-289(UAG), his3Δ-200, ura3-52, HSP104::LEU2) (kindly provided by S. Lindquist, Whitehead Institute of Biomedical Research, USA) were used in the current work. The yeast cells were maintained on the YEPD medium (yeast extract, 5 g/liter; peptone, 10 g/liter; glucose, 20 g/liter and agar-agar, 15 g/liter) at 30°C. Yeast cells were grown for 14-16 h at 30°C in 100 ml flasks containing 25 ml of liquid medium YEPD. For the experiments, a certain amount of 14-h culture was inoculated into fresh medium and incubated until reaching a concentration of 2·107 cells/ml.
Isolation of total protein and Western blotting. For isolation of total protein, the yeast cells were pelleted by centrifugation, washed, and stored at –70°C until the isolation of the protein. Before protein extraction the cells were thawed, resuspended in isolation buffer (0.1 M Tris-HCl, 3 mM SDS, 1 mM β-mercaptoethanol, pH 7.4-7.6), frozen in liquid nitrogen, and ground with quartz powder. Crude cellular components were removed by centrifugation (15,000g, 15 min), and the protein was precipitated by a threefold volume of cold acetone. The protein precipitate was washed three times with acetone and dissolved in sample buffer (0.625 M Tris-HCl, 8 mM SDS, 0.1 M β-mercaptoethanol, 10% glycerol, and 0.001% ethyl bromophenol blue, pH 6.8). Protein concentration was determined by the method of Lowry et al. [19]. Following SDS-PAGE in 12% polyacrylamide gel, the gel was immunoblotted with antibodies against anti-Hsp104 (SPA-8040; StressGen, USA) and Hsp60 (US Biological H1830-77B, USA) according to a previously reported method [20].
Counting of colony-forming units (CFU). To count CFU, the yeast cells were diluted and plated in a standard way in YEPD medium containing 1.5% agar. After 24-48 h incubation at 30°C, the CFU were counted, and the data are represented as a percentage with respect to control in three independent experiments.
Determination of reactive oxygen species (ROS). To determine ROS generation, the fluorescent dye 2′,7′-dichlorodihydrofluorescein diacetate H2DCF·DA was used at final concentration of 50 µM. The results were recorded after incubation of the cells with the dye for 10 min (50°C) or 60 min (45°C). Fluorescence microscopy was carried out using an AxioObserverZ1 inverted fluorescence microscope (Germany) with digital monochrome camera AxioCamMRm3 and software package AxioVisionRel.4.6.
All experiments were repeated a minimum of three times. The data were analyzed statistically, i.e. arithmetic means and standard errors were determined.
RESULTS
Pretreatment by cycloheximide increased basal thermotolerance of yeast cells. Treatment at 45°C is a minimal heat regime causing a significant decrease in viability of S. cerevisiae cells during 60 min of exposure [21]. Treatment at 50°C is a standard regime commonly used in experiments to study the mechanism of induced thermotolerance [2]. Therefore, treatments at 45 and 50°C were used to study the effect of cycloheximide on thermotolerance depending on the intensity. Cells were treated during 30 min at 30°C in the presence of 20 µg/ml cycloheximide, then washed free the from drug, resuspended in fresh medium, and subjected to heat shock as indicated (Fig. 1a). It was previously shown that cycloheximide under these conditions effectively inhibited de novo protein synthesis (Fig. 2b).
Fig. 1. Effect of cycloheximide on basal thermotolerance. Saccharomyces cerevisiae cells (strain Ψ-74-D694) grown in YEPD were treated at 30°C (30 min) in the presence of 20 µg/ml cycloheximide (CHM) (2) or without it (1). After washing free from the agent, the cells were exposed to heat shock at 45 or 50°C. Survival was evaluated by CFU counting after 48 h of incubation at 30°C. a) Scheme of experiment; b, c) CFU counts after exposure to heat shock 45 and 50°C, respectively; n = 3, mean ± SE. HS, heat shock (45 or 50°C).
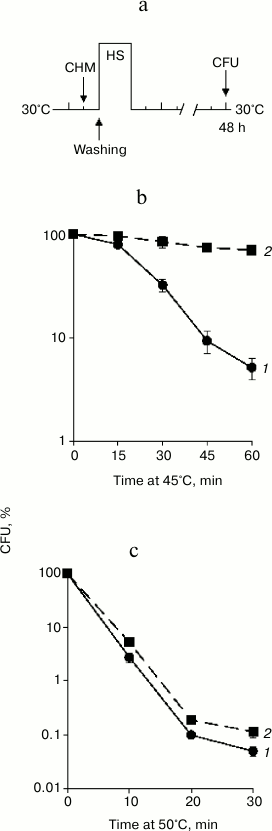
As expected, treatment at 45°C resulted in a gradual decrease in viability (Fig. 1b). Severe heat shock at 50°C produced a more significant negative effect on viability. Nevertheless, the percentage of viable cells after 10 min of treatment at 50°C and after 60 min of treatment at 45°C was approximately the same (Fig. 1c). The study of the effect of cycloheximide on thermotolerance showed that pretreatment with cycloheximide at 30°C strongly protected S. cerevisiae cells from death in the case of moderate heat shock (Fig. 1b). Under conditions of severe heat shock, pretreatment with cycloheximide also exerted a protective effect, but to a far less extent (Fig. 1c). For instance, cycloheximide increased the viability of the yeast after treatment at 45°C (60 min) more than 10-fold (Fig. 1b), while after the heat shock at 50°C (10 min) the number of viable cells treated with cycloheximide was only twice as high as those in the control (Fig. 1c). Consequently, the effect of cycloheximide on yeast thermotolerance depended on the intensity of the heat exposure. Cycloheximide effectively suppressed yeast cells death induced by moderate heat shock (45°C), but the protective effect was slight under conditions of severe heat shock (50°C).
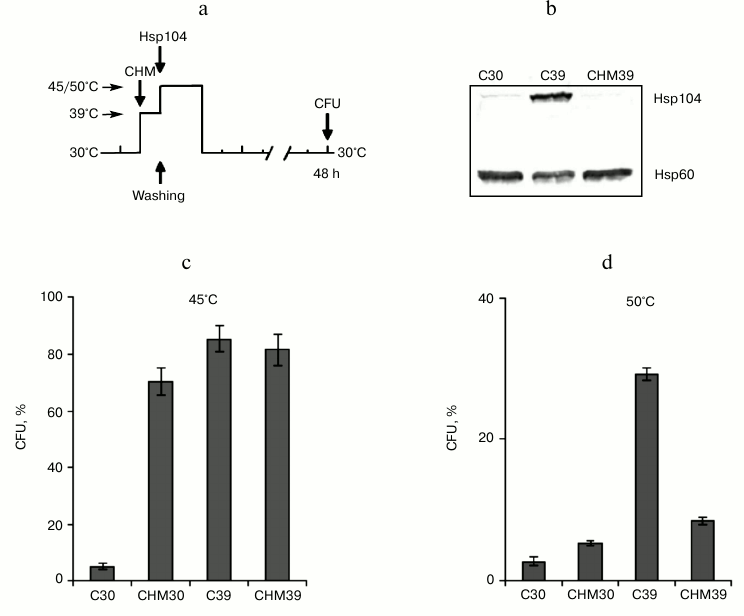
Effect of cycloheximide on induced thermotolerance in yeast cells depends on intensity of heat shock. Pretreatment at 39°C (30 min) induced the synthesis of Hsp104 (Fig. 2b), which was accompanied by the development of induced thermotolerance of yeast cells to moderate (Fig. 2c) and severe (Fig. 2d) heat shock. To examine the effect of cycloheximide on induced thermotolerance in dependence on intensity of heat shock, the yeast cells were incubated at 30 or 39°C in the presence of cycloheximide, as described above, and treated at 45 or 50°C (Fig. 2a). As follows from Fig. 2 (c and d), cycloheximide affected the ability of preliminary heat stress at 39°C to induce thermotolerance to the damaging heat shock at 45 and 50°C in different ways. Cycloheximide did not suppress induced thermotolerance when cells were exposed to moderate heat shock at 45°C (Fig. 2c). However, cycloheximide significantly inhibited this process if the cells were subjected to severe heat shock at 50°C (Fig. 2d). It is worth noting that under conditions of moderate heat shock the pretreatment with cycloheximide at 30°C (CHM30) induced the yeast thermotolerance approximately to the same extent as the preliminary heat stress at 39°C (C39) (Fig. 2c). However, the heat stress (C39) was much more effective than cycloheximide (CHM30) in protecting yeast cells from death under conditions of severe heat shock (Fig. 2d). Thus, treatment with cycloheximide effectively suppressed the induction of Hsp104 synthesis during heat stress at 39°C (Fig. 2b) and, at the same time, suppressed the induced thermotolerance to severe heat shock at 50°C (Fig. 2d), but it produced no effect on induced thermotolerance to moderate heat shock at 45°C (Fig. 2c). Therefore, the ability of cycloheximide to suppress induced thermotolerance in yeast cells depended on the intensity of the heat exposure.
Fig. 2. Effect of cycloheximide on induced thermotolerance and Hsp104 synthesis. Saccharomyces cerevisiae cells (strain Ψ-74-D694) grown in YEPD were treated at 30 or 39°C (30 min) in the presence of 20 µg/ml cycloheximide (CHM30, CHM39) or without it (C30, C39). After washing free from the agent, the cells were exposed to heat shock at 45 or 50°C or used for Hsp104 and Hsp60 assays. Survival was evaluated by CFU counting after 48 h of incubation at 30°C. a) Scheme of experiment; b) synthesis of Hsp104 and Hsp60 at 30°C (C30), 39°C (C39) and 39°C in the presence of cycloheximide (CHM39); c, d) CFU counts after exposure to heat shock at 45°C (60 min) and 50°C (10 min), respectively; n = 3, mean ± SE.
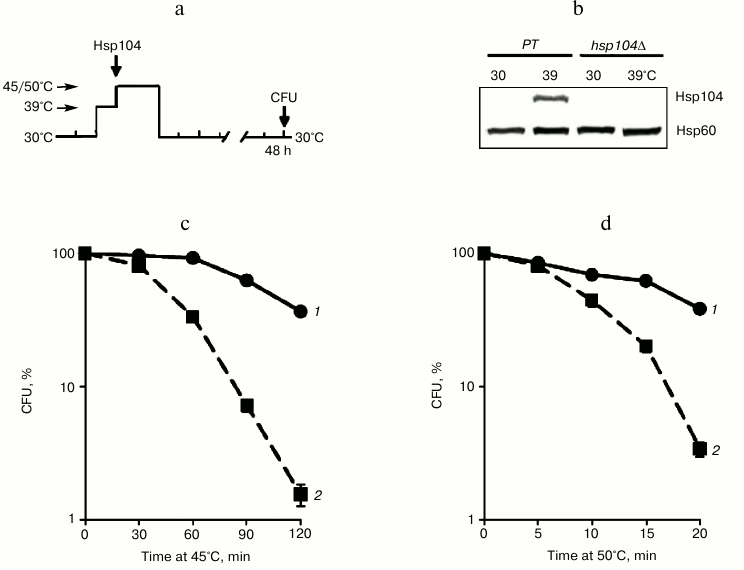
Hsp104 protected yeast cells from death under moderate and severe heat shock conditions. Since cycloheximide effectively inhibited the induction of Hsp104 synthesis during heat stress at 39°C (Fig. 2b), but it did not reduce the ability of the yeast to develop thermotolerance to 45°C, one can expect that Hsp104 does not have a protective effect on yeast viability under conditions of moderate heat exposure. To test this hypothesis, the hsp104Δ mutant was used, which does not synthesize Hsp104 during heat stress (Fig. 3b). Cells of the hsp104Δ mutant and parent type were treated at 39°C (30 min) (Fig. 3a) to induce Hsp104 synthesis (Fig. 3b) and then subjected to heat exposure at 45 and 50°C. As follows from Figs. 3c and 3d, the hsp104Δ mutant cells lost viability much more severely after moderate (45°C) and severe (50°C) heat treatment compared to the parent type cells. Therefore, Hsp104 protects cells against the lethal heat shock independently of its intensity.
Fig. 3. Effect of Hsp104 on thermotolerance to severe and moderate heat shock. Saccharomyces cerevisiae cells (parent type (PT) strain Ψ-74-D694 and isogenic hsp104Δ mutant) grown in YEPD were treated at 39°C (30 min). Then the cells were exposed to heat shock of 45 or 50°C or used for Hsp104 and Hsp60 assays. Survival was evaluated by CFU counting after 48 h of incubation at 30°C. a) Scheme of experiment; b) synthesis of Hsp104 and Hsp60; c, d) CFU counts of parent type (1) and mutant (2) cells after exposure to heat shock 45 and 50°C, respectively; n = 3, mean ± SE.
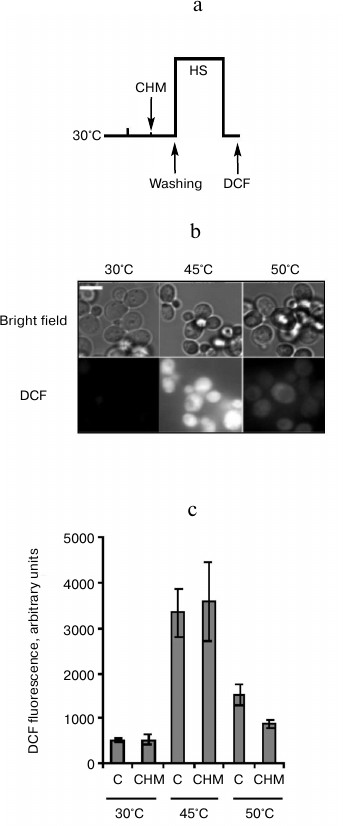
Effect of cycloheximide on ROS generation induced in yeast cells by heat shock. Cellular protein denaturation and aggregation as well as an increase in ROS generation is thought to be the primary cause of heat-induced cell death [2]. An increase in ROS generation is a known trigger of PCD [11]. Therefore, the in next experiment the effect of cycloheximide on ROS generation induced by moderate and severe heat shock was examined (Fig. 4a). ROS generation was determined by measuring DCF fluorescence. As follows from Fig. 4b, under ordinary incubation conditions (30°C) DCF fluorescence was almost undetected. Moderate heat shock (45°C, 60 min) resulted in a sharp increase in DCF fluorescence. Severe heat shock (50°C, 10 min) also led to an increase in fluorescence, but the intensity of fluorescence was significantly less compared to the moderate heat shock (Fig. 4b). Pretreatment with cycloheximide produced an ambiguous effect on DCF fluorescence at elevated temperatures. There was no effect of cycloheximide on increase in DCF fluorescence after treatment at 45°C, but cycloheximide suppressed DCF fluorescence after treatment at 50°C (Fig. 4c).
Fig. 4. Effect of cycloheximide on ROS generation induced by heat shock. Saccharomyces cerevisiae cells (strain Ψ-74-D694) grown in YEPD were treated at 30°C (30 min) in the presence of 20 µg/ml cycloheximide (CHM) or without it (C). After washing free from the agent, the cells were exposed to heat shock at 45 (60 min) or 50°C (10 min). DCF fluorescence was measured immediately after treatment. a) Scheme of experiment; b) microphotographs of cells. Bar, 5 µm. c) Counting of DCF fluorescence, n = 4, mean ± SE.
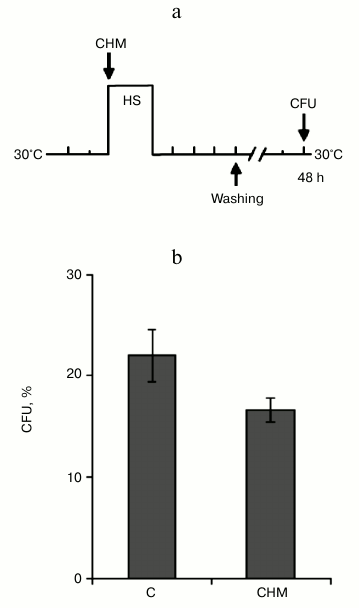
Protective effect of cycloheximide on yeast cell viability did not depend on inhibition of pro-PCD protein synthesis. In previous experiments, treatment by cycloheximide was performed during 30 min at 30°C, and then cells were washed and subjected to lethal heat shock (Figs. 1a and 2a). PCD is an active process and requires the activation of proapoptotic (or pro-PCD) proteins, which, in fact, initiated a cell suicide program. Therefore, it was assumed that cycloheximide suppresses PCD development and, respectively, enhances the yeast viability due to the ability of the drug to inhibit pro-PCD protein synthesis [16, 17]. Therefore, the aim of the following experiment was to investigate whether or not the ability of cycloheximide to increase yeast thermotolerance under conditions of moderate heat shock (Fig. 1b) is due to inhibition of synthesis of putative pro-PCD proteins. Theoretically, activation of the pro-PCD protein synthesis should be expected at the start of stress exposure or immediately after it. Therefore, the yeast cells were treated with cycloheximide immediately before heat shock, and then incubated further in the presence of the agent during moderate heat shock (45°C, 60 min) and subsequent recovery period at 30°C, 240 min (Fig. 5a). Under these experimental conditions, the protective effect of cycloheximide on yeast viability was absent. Moreover, cycloheximide slightly but reproducibly reduced the viability of the yeast (Fig. 5b). These results indicate that the protective effect of cycloheximide on yeast viability under conditions of moderate heat shock (Figs. 1b and 2c) was not dependent on inhibition of the synthesis of pro-PCD proteins.
Fig. 5. Combined effect of cycloheximide and heat shock on thermotolerance. Saccharomyces cerevisiae cells (strain Ψ-74-D694) grown in YEPD were incubated for 60 min at 45°C and 240 min at 30°C in the presence of 20 µg/ml cycloheximide (CHM) or without it (C), then washed free from the agent. Survival was evaluated by CFU counting after 48 h of incubation at 30°C. a) Scheme of experiment; b) CFU counts after exposure to heat shock.
DISCUSSION
Mechanism of heat shock-induced yeast cell death depends on intensity of heat exposure. Cycloheximide treatment inhibited the yeast cell death induced by low concentrations of acetic acid [17] and hydrogen peroxide [16], but death induced by high concentrations of these agents was not suppressed by cycloheximide. Cycloheximide produced a similar effect on yeast cell death under heat shock conditions. Pretreatment with cycloheximide effectively suppressed cell death induced by moderate heat shock (Fig. 1b) and produced a slight protective effect during severe heat shock (Fig. 1c). The prevention of cell death by cycloheximide serves as a specific criterion to differentiate PCD from necrosis [16-18]. According to this criterion, the results of the current study indicate that moderate heat shock at 45°C activated PCD program in yeast cells. Heat shock at 42°C was shown to induce PCD in cultured mammalian cells as evidenced by caspase activation. Caspase activation was not observed during severe heat shock at 46°C, and the cells died by necrosis [10]. A similar situation was probably observed in yeast cells: moderate heat shock activated the development of PCD, and the process was inhibited by cycloheximide. Under conditions of severe heat shock, the cells lost their viability mainly as a result of necrosis, which was not affected by cycloheximide. This assumption is corroborated by measurements of ROS generation under moderate and severe heat shock conditions. Increased ROS production is one of the indicators of PCD development [11]. Heat shock at 45°C (60 min) and at 50°C (10 min) produced approximately the same negative effect on yeast viability (Fig. 2). However, the intensity of ROS production under moderate heat shock was approximately two times higher in comparison to severe heat shock (Fig. 4). Therefore, the mechanism of yeast cell death appears to depend on the intensity of heat exposure.
These results are consistent with work [14]. Sharp temperature elevation to 52°C was shown to cause yeast cell death that is not dependent on Dnm1 and Yca1, known activators of PCD. However, ramped elevation of temperature to 51°C (within 15 min) induced cell death that was suppressed by DNM1 and YCA1 deletions [14].
Cycloheximide effect on induced thermotolerance in yeast cells depends on mechanism of heat-induced cell death. Heat stress at 39°C caused development of induced thermotolerance in S. cerevisiae cells under moderate and severe heat shock conditions (Fig. 2). Cycloheximide produced no negative effect on this phenomenon if the cells were heat shocked at 45°C (Fig. 2c). In contrast, cycloheximide significantly suppressed development of induced thermotolerance if the cells were treated at 50°C (Fig. 2d). Obviously, cycloheximide did not inhibit induced thermotolerance to moderate heat shock, because pretreatment by this agent produced a significant protective effect under these conditions (Fig. 2c). However, cycloheximide effectively suppressed induced thermotolerance to severe heat exposure, while pretreatment with the drug did not result in significant protection (Fig. 2d). This result helps to resolve the conflicting results of cycloheximide effects on induced thermotolerance previously obtained by other researchers [3-7]. The probable reason for the contradictions is the ability of cycloheximide to inhibit PCD induced by heat shock. This fact has not been previously taken into account. Thus, whether or not cell death proceeds by the PCD pathway, it seems to depend on the intensity of heat exposure, yeast strain used, and experimental conditions. So it is not surprising that in one experimental system cycloheximide prevented the development of induced thermotolerance, while in another experimental system it had no effect.
Hsp104 as a negative regulator of PCD in yeast cells. It is well known that induction of Hsp104 synthesis in yeast cells plays a crucial role in induced thermotolerance [2, 8, 9]. However, despite the fact that cycloheximide inhibited induction of Hsp104 synthesis during mild heat stress (Fig. 2b), the development of induced thermotolerance to moderate heat shock was not suppressed (Fig. 2c). The question arises – does the ability of Hsp104 to perform its functions depend on the mechanism of cell death induced by heat shock? A comparison of the development of induced thermotolerance in the parent type strain and hsp104Δ mutant showed that loss of Hsp104 reduced induced thermotolerance in yeast cells subjected to severe and moderate heat shock to a similar extent (Fig. 3). The prevention of cell death as the result of cycloheximide treatment and increased generation of ROS are regarded as specific indicators of PCD [11, 16, 17]. Since cell death during temperature elevation up to 45°C was accompanied by increased ROS generation (Fig. 4) and was effectively suppressed by cycloheximide (Fig. 1b), the data indicate that Hsp104 inhibits PCD development in S. cerevisiae cells during moderate heat shock (Fig. 3c). This concept is confirmed by expression of Hsp104 S. cerevisiae in human cells. An increase in Hsp104 expression inhibited caspase-3 activation and thereby suppressed PCD during heat shock [22]. It is possible that Hsp104 inhibits development of PCD via interaction with metacaspase Yca1 [23] or components of the actin cytoskeleton [24]. Activation of metacaspase Yca1 and disruption of actin cytoskeleton are putative PCD triggers in yeast cells [11]. Probably not only Hsp104, but also other yeast Hsps could modulate PCD development. There are four genes SSA1-SSA4 (stress seventy subfamily A) encoding cytosolic members of the yeast Hsp70 family [2, 9]. An increase in Ssa3 inhibited PCD development in yeast cells expressing α-synuclein, a trigger of neurodegenerative diseases in humans [25]. SSA1 deletion stimulated PCD induced by acetic acid. Interestingly, on the contrary SSA2 deletion suppressed PCD [14].
Protective effect of cycloheximide on yeast thermotolerance could not be explained by suppression of synthesis of pro-PCD proteins. PCD development was assumed to be determined by synthesis of putative pro-PCD proteins, respectively, cycloheximide inhibits PCD by suppressing their synthesis [16, 17]. Analysis of protein expression profiles showed that in S. cerevisiae cells during PCD induced by H2O2 treatment, the amounts of triosephosphate isomerase and glyceraldehyde-3-phosphate dehydrogenase were increased [26]. However, the protective effect of cycloheximide on thermotolerance could not be explained by inhibition of synthesis of putative pro-PCD proteins. The induction of protein synthesis activating PCD should be expected at the start of heat exposure or immediately after it. However, the protective effect of cycloheximide was observed only in the case of pretreatment at 30°C (Fig. 1c). The protective effect was absent if cycloheximide was added immediately before heat exposure and cells were incubated with the drug during the shock and subsequent recovery period (Fig. 5).
Inhibition of general protein synthesis is a protective reaction of yeast cells to heat shock. The results suggest that the reason for the protective effect of cycloheximide is the inhibition of general protein synthesis. Cellular protein denaturation and aggregation is thought to be the primary cause of heat shock-induced cell death [2]. As newly synthesized proteins undergoing the process of folding are supposed to be particularly prone to thermal denaturation [2, 5], it could be advantageous for cells not to continue translation under conditions under which nascent polypeptides will be anyway damaged by denaturing conditions and subject to aggregation [2]. Indeed, the heat stress protecting cells from death leads not only to induction of Hsps synthesis, but it also inhibits general protein synthesis [27]. From this point of view, the natural inhibition of general protein synthesis and induction of Hsp synthesis by a mild heat stress should be considered as independent adaptive reactions preventing newly synthesized polypeptides from denaturation and aggregation during subsequent more severe heat shock. Therefore, if induced thermotolerance depends on a combination of Hsp induction and inhibition of general protein synthesis, then the former process should be antagonized by cycloheximide treatment, while the latter process should not, as cycloheximide would act in the same direction. This supposition explains why there was no effect of cycloheximide on induced thermotolerance during moderate heat shock (Fig. 2c) despite the fact that cycloheximide effectively suppressed heat-induced Hsp104 synthesis (Fig. 2b). More severe heat shock is assumed to induce the denaturation and aggregation of not only newly synthesized proteins, but also proteins that are in their native state. Under these conditions, the cell had no tools to withstand heat shock if Hsp synthesis during mild heat stress was blocked by cycloheximide (Fig. 2d). In general, the results indicate that at least two independent mechanisms of induced thermotolerance are operating in the cells under moderate heat shock: inhibition of general protein synthesis and induction of Hsp synthesis. Both mechanisms are aimed to prevent the formation of protein aggregates. The effective inhibition of protein synthesis under these conditions significantly protects cells from death despite blocking Hsps synthesis. During severe heat shock the induced thermotolerance depends mainly on the induction of Hsp synthesis, and suppression of induction blocks the development of thermotolerance.
It could be assumed that the ability of cycloheximide to inhibit PCD development in yeast cells induced by hydrogen peroxide [16], acetic acid [17], and α-factor [18] are also determined by inhibition of general protein synthesis. Growth rate depends on the intensity of protein synthesis. A decrease in yeast growth rate is shown to lead to cell resistance to heat shock [28], as well as to hydrogen peroxide and acetic acid treatment [29].
Denatured and aggregated proteins as PCD triggers. The main factors determining PCD in yeast cells are the redistribution of phosphatidylserine in the cytoplasmic membrane, increased ROS production, chromatin condensation, and DNA fragmentation [11]. The appearance of aggregated proteins is probably an additional factor affecting the development of PCD [13, 15, 30]. Metacaspase Yca1 is required for clearance of insoluble protein aggregates [23]. On this basis, the denaturation and aggregation of nascent proteins was assumed to stimulate PCD in yeast cells, and cycloheximide blocks this process by inhibiting the synthesis of aggregation-prone proteins. The absence of any cycloheximide effect on ROS production in yeast cells in the case of moderate heat shock (Fig. 4c) indicates that the appearance of aggregated proteins is not the cause of oxidative stress under these conditions. Rather, increased level of ROS contributes to appearance of aggregated proteins. Oxidation of proteins leads to their carbonylation. Carbonylated proteins tend to form aggregates [2]. Consequently, Hsp104 and cycloheximide via prevention of aggregation and denaturation of newly synthesized cellular proteins could block the development of PCD in yeast cells under condition of moderate heat shock. To consider aggregated proteins as one of the triggers of PCD, it is interesting to note that both Hsp104 [2, 9] and Yca1 [23] interact with aggregated proteins and facilitate their removal from the cell. The question arises, why are proteins with similar functions likely to play an opposite role in PCD?
This study was supported by the Ministry of Education and Science of Russian Federation, project 8266.
REFERENCES
1.Alexandrov, V. J., and Kislyuk, I. M. (1994)
Tsitologiya, 36, 5-59.
2.Verghese, J., Abrams, J., Wang, Y., and Morano, K.
A. (2012) Microbiol. Mol. Biol. Rev., 76,
115-158.
3.McAlister, L., and Finkelstein, D. B. (1980)
Biochem. Biophys. Res. Commun., 93, 819-824.
4.Craig, E. A., and Jacobsen, K. (1984) Cell,
38, 841-849.
5.Piper, P. W. (1993) FEMS Microbiol. Rev.,
11, 339-355.
6.Hall, B. G. (1983) J. Bacteriol.,
156, 1363-1365.
7.Watson, K., Dunlop, G., and Cavicchioli, R. (1984)
FEBS Lett., 172, 299-302.
8.Rikhvanov, E. G., Varakina, N. N., Rusaleva, T. M.,
Rachenko, E. I., Knorre, D. A., and Voinikov, V. K. (2005) Curr.
Genet., 48, 44-59.
9.Rikhvanov, E. G., Romanova, N. V., and Chernoff, Y.
O. (2007) Prion, 4, 217-222.
10.Samali, A., Holmberg, C. I., Sistonen, L., and
Orrenius, S. (1999) FEBS Lett., 461, 306-310.
11.Sukhanova, E. I., Rogov, A. G., Severin, F. F.,
and Zvyagilskaya, R. A. (2012) Biochemistry (Moscow), 77,
761-775.
12.Chen, S. R., Dunigan, D. D., and Dickman, M. B.
(2003) Free Radic. Biol. Med., 34, 1315-1325.
13.Fahrenkrog, B., Sauder, U., and Aebi, U. (2004)
J. Cell Sci., 117, 115-126.
14.Teng, X., Cheng, W. C., Qi, B., Yu, T. X.,
Ramachandran, K., Boersma, M. D., Hattier, T., Lehmann, P. V., Pineda,
F. J., and Hardwick, J. M. (2011) Cell Death Dis., 4,
e188.
15.Cebulski, J., Malouin, J., Pinches, N., Cascio,
V., and Austriaco, N. (2011) PLoS One, 6,
e20882.
16.Madeo, F., Frohlich, E., Ligr, M., Grey, M.,
Sigrist, S. J., Wolf, D. H., and Frohlich, K. U. (1999) J. Cell
Biol., 145, 757-767.
17.Ludovico, P., Sousa, M. J., Silva, M. T., Leao,
C., and Corte-Real, M. (2001) Microbiology, 147,
2409-2415.
18.Severin, F. F., and Hyman, A. A. (2002) Curr.
Biol., 12, 233-235.
19.Lowry, O. H., Rosebrough, N. J., Farr, A. L., and
Randall, R. J. (1951) J. Biol. Chem., 193, 265-275.
20.Timmons, T. M., and Dunbar, B. S. (1990)
Methods Enzymol., 182, 679-701.
21.Fedoseeva, I. V., Pjatricas, D. V., Varakina, N.
N., Rusaleva, T. M., Stepanov, A. V., Rikhvanov, E. G., Borovskii, G.
B., and Voinikov, V. K. (2012) Biochemistry (Moscow), 77,
78-86.
22.Mosser, D. D., Ho, S., and Glover, J. R. (2004)
Biochemistry, 43, 8107-8715.
23.Lee, R. E., Brunette, S., Puente, L. G., and
Megeney, L. A. (2010) Proc. Natl. Acad. Sci. USA, 107,
13348-13353.
24.Tessarz, P., Schwarz, M., Mogk, A., and Bukau, B.
(2009) Mol. Cell. Biol., 29, 3738-3745.
25.Flower, T. R., Chesnokova, L. S., Froelich, C.
A., Dixon, C., and Witt, S. N. (2005) J. Mol. Biol., 351,
1081-1100.
26.Magherini, F., Tani, C., Gamberi, T., Caselli,
A., Bianchi, L., Bini, L., and Modesti, A. (2007) Proteomics,
7, 1434-1445.
27.Gasch, A. P., Spellman, P. T., Kao, C. M.,
Carmel-Harel, O., Eisen, M. B., Storz, G., Botstein, D., and Brown, P.
O. (2000) Mol. Biol. Cell, 11, 4241-4257.
28.Rikhvanov, E. G., Varakina, N. N., Rusaleva, T.
M., Rachenko, E. I., and Voinikov, V. K. (2003) Microbiology,
72, 423-428.
29.Zakrzewska, A., van Eikenhorst, G., Burggraaff,
J. E., Vis, D. J., Hoefsloot, H., Delneri, D., Oliver, S. G., Brul, S.,
and Smits, G. J. (2011) Mol. Biol. Cell, 22,
4435-4446.
30.Sokolov, S., Pozniakovsky, A., Bocharova, N.,
Knorre, D., and Severin, F. (2006) Biochim. Biophys. Acta,
1757, 660-666.