Differential Binding of Plasma Proteins by Liposomes Loaded with Lipophilic Prodrugs of Methotrexate and Melphalan in the Bilayer
N. R. Kuznetsova and E. L. Vodovozova*
Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia; fax: +7 (495) 330-6601; E-mail: elvod@lipids.ibch.ru* To whom correspondence should be addressed.
Received March 26, 2014; Revision received April 23, 2014
Immediately upon contact with blood, nanosized drug delivery systems become coated with a so-called protein corona. The quantitative and qualitative composition of the corona defines not only the behavior of the nanocarrier in the circulation but, ultimately, the pharmacokinetics and biodistribution of the encapsulated drug as well. In turn, the composition of the protein corona depends on the surface properties of the nanoparticles, such as size and distribution of charge and functional groups on the particle surface. Liposomes belong to the most bio- and hemocompatible drug delivery systems feasible for intravenous route of administration required in chemotherapy of metastasizing tumors. However, knowledge on the interactions of liposomes of various compositions with blood plasma proteins remains fragmentary. Moreover, all nanosized drug delivery systems are potential targets for the innate immunity system, primarily the complement (C) system, which underlies frequent cases of hypersensitivity reactions. Recently, in a panel of in vitro hemocompatibility tests, we demonstrated that liposomes built of natural phospholipids – egg phosphatidylcholine and phosphatidylinositol from Saccharomyces cerevisiae – and loaded with diglyceride conjugates of anticancer drugs melphalan and methotrexate, did not affect the morphology and numbers of the main blood cell types. While preparations with melphalan prodrug were also inert in coagulation and C activation tests, methotrexate-loaded liposomes caused impaired coagulation and C activation. The aim of this work was to study the interactions of liposomes carrying prodrugs of melphalan and methotrexate with blood plasma proteins in vitro. Data on protein binding capacity of liposomes obtained with classical gel permeation chromatography techniques allowed for prediction of rather rapid elimination of the liposomes from circulation. A number of differences revealed through immunoblotting of the liposome-bound proteins agree with the previously obtained data on C activation. The possible mechanism of C activation by methotrexate-containing liposomes is discussed.
KEY WORDS: nanomedicine, antitumor liposomes, melphalan, methotrexate, lipophilic prodrugs, protein corona, complementDOI: 10.1134/S0006297914080070
Abbreviations: C, complement; Mlph-DOG, diglyceride conjugate of melphalan; Mlph-L, liposomes loaded with Mlph-DOG; MTX-DOG, diglyceride conjugate of methotrexate; MTX-L, liposomes loaded with MTX-DOG; PB, protein binding; PBS, phosphate buffered saline; PC, phosphatidylcholine; PI, phosphatidylinositol.
Immediately upon contact with biological media, primarily the blood
plasma, nanosized particles become covered with proteins and their
complexes with lipid molecules (lipoproteins) in the same manner as do
macromaterials [1-4]. The
so-called protein corona modifies the physicochemical properties of the
drug carrier surface. The effective unit of delivery is not the
nanoparticle as such, but the complex it forms with proteins more or
less tightly bound to its surface. It is the corona of liposomes that
interacts with cell receptors in the organism; therefore, the
qualitative and quantitative composition of the adsorbed proteins
determines biodistribution of the encapsulated drug and its
pharmacokinetics. In turn, the composition of the protein corona
depends on the properties of the liposome surface, which is
predetermined by liposome size and distribution of charge and
functional groups on the surface. Variation of these parameters may
only modify the amount and composition of proteins bound by the
particle, but it cannot eliminate the possibility of binding (e.g. see
[5]).
Traditionally, interactions of liposomes with blood plasma proteins have been investigated in the context of opsonization of the liposome surface and the related problem of preliminary withdrawal of liposomes from the circulation by cells of the reticuloendothelial system [6]. The laws of liposome behavior in biological media unveiled to date are complemented by knowledge gained for other nanosized drug delivery systems (polymer micelles, dendrimers, polyelectrolyte complexes, etc.), and researchers attempt to summarize the generated body of data [3]. However, the picture of the interactions of liposomes with proteins remains fragmentary. In this connection, special study on the behavior of new liposomal preparations upon their contact with blood plasma is of immediate interest.
Liposomes, as well as other supramolecular drug delivery systems, although built from inherently non-immunogenic molecules, resemble pathogenic viruses in their dimensions and surface structure presenting molecular templates and repeated elements recognized by receptors of immune cells. Therefore, nanosized carriers are potential targets of the innate immune system, first of all, the complement (C) system [7]. After the first nanomedicines had been approved in clinics, a wide range of data has been collected on hypersensitivity reactions observed in response to intravenous administration of such preparations as Doxil® (liposomes stabilized by surface-grafted polyethylene glycol chains, the so-called PEGylated liposomes, with an antitumor antibiotic doxorubicin in the inner volume) [8] and Taxol® (emulsion of a cytostatic paclitaxel stabilized with the pharmacopeial detergent Cremophor EL) (e.g. [9]).
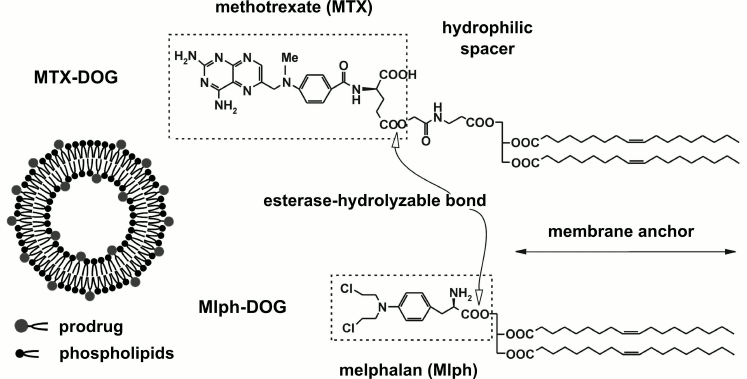
To improve the pharmacological properties of chemotherapy drugs, we suggest that they should be incorporated into the lipid bilayer of 100-nm liposomes in the form of lipophilic prodrugs, namely, ester conjugates with lipids [10-12]. In a panel of in vitro hemocompatibility tests, liposomes composed of natural phospholipids loaded with diglyceride conjugates of melphalan and methotrexate (Mlph-DOG and MTX-DOG, respectively; see Fig. 1) did not affect the main blood cells [13]. However, in contrast to the inert formulations of the melphalan prodrug, methotrexate-loaded liposomes induced impairment of blood coagulation and activation of the C system, as evidenced by the production of C3a anaphylatoxin detected by ELISA and exhaustion of the whole system in the serum residual hemolytic activity test [13].
Fig. 1. Structures of lipophilic prodrugs Mlph-DOG and MTX-DOG and schematic representation of the liposome.
Obviously, the malfunction of coagulation and complement cascades is caused by liposome interaction with protein factors and resulting decrease in the available amount of intact proteins in plasma. Concentrations of various coagulation factors in plasma are very low; for example, factors V, VII (in activated plasma), and X are present at concentrations of 20, 10, and 200 nM, respectively. At the same time, concentration of fibrinogen, one of the major plasma proteins, is 9 µM [14]. Therefore, even a slight (non)specific sorption of one of the coagulation factors on the surface of liposomes may lead to a considerable shift in the cascade reaction equilibria.
The aim of this work was to study the interactions of liposomes loaded with Mlph-DOG and MTX-DOG with blood plasma proteins in vitro. First, we determined the simplest quantitative characteristic of the interaction, namely the capacity of liposomes for protein binding (PB). PB value may be a prognostic factor for comparative evaluation of circulation times for liposomes of new lipid compositions [4, 6]. Then, immunoblotting of liposome-bound proteins revealed a number of differences between the liposome formulations, which allowed for a hypothesis on how methotrexate-containing liposomes activated complement in vitro [13].
MATERIALS AND METHODS
Diglyceride conjugates of melphalan [15] and methotrexate [16] were synthesized as previously described. Egg yolk phosphatidylcholine (PC) and phosphatidylinositol (PI) from Saccharomyces cerevisiae were from Reakhim (Russia), Sepharose CL-4B from Pharmacia (Sweden), and EDTA and the rest of the reagents were purchased from Sigma or Flow Laboratories (USA). Solvents were purified using standard methods; evaporation was performed at temperatures below 40°C. Phosphate-buffered saline (PBS) supplemented with 1 mM EDTA, pH 7.1, contained KH2PO4, 0.2 g/liter; NaH2PO4·2H2O, 0.15 g/liter; Na2HPO4, 1.0 g/liter; KCl, 0.2 g/liter, and NaCl, 8.0 g/liter.
Liposome preparation. Liposomes (L) were prepared from mixtures of PC–PI–MTX-DOG/Mlph-DOG, 8 : 1 : 1 (molar ratio) by extrusion of the lipid dispersions through calibrated nuclear pore filters as previously described [10, 13]. Briefly, a mixed solution of lipids and a prodrug in chloroform was evaporated in a round-bottom flask on a rotary evaporator (Heidolph, Germany). Typically, the mixtures contained 12.5 mg PC, 1.6 mg PI, and 2.4 mg (2 µmol) MTX-DOG or 1.8 mg (2 µmol) Mlph-DOG. The lipid films were further dried for 30 min at 5 Pa and then hydrated during 2 h at room temperature in 0.5 ml PBS. The suspension was shaken and subjected to five cycles of freezing and thawing (liquid N2/40°C) and extruded 20 times through polycarbonate membrane filters (Nucleopore, USA) with pore diameter of 100 nm using an Avanti mini-extruder (Avanti Polar Lipids, USA). Concentrations of the prodrugs in dispersions were determined after destruction of the liposomes with ethanol: UV spectra were registered, and optical density was measured at the absorption maxima (MTX-DOG: λmax 302 nm, ε ~ 25,000 M–1·cm–1; Mlph-DOG: λmax 258 nm, ε ~ 19,700 M–1·cm–1) on an SF-256-UVI (LOMO Fotonika, Russia) double-beam spectrophotometer. Particle size was assessed by dynamic light scattering using a Brookhaven Particle Analyzer 90+ (USA). The average size of the liposomes with Mlph-DOG (Mlph-L) was 90.4 ± 1.0 nm, and that of MTX-DOG (MTX-L) was 89.7 ± 1.3 nm (mean diameter values for two independent batches of liposomes).
Incubation of liposomes with plasma and isolation of liposome–protein complexes. Blood of healthy donors was collected into Terumo Venosafe (Terumo Europe N. V., Belgium) tubes over citrate. Plasma was separated by centrifugation at 2000g for 5 min. Joint plasma of four donors was used. Liposomes (90 µl) were incubated with 360 µl plasma at 37°C and weak shaking in Eppendorf (Germany) tubes (1.5 ml) for 15 min unless stated otherwise. The mixture was applied to a Sepharose CL-4B column (~1.1 × 16 cm; V0 = 7 ml) and eluted with PBS; fractions of 400 µl were collected. Ethanol (400 µl) was added to aliquots of fractions (80 µl), the mixture was centrifuged for 10 min at 10,000 rpm, and the supernatants were analyzed for the prodrug content by spectrophotometry. In parallel, 100 µl of each fraction was collected for protein determination. In some experiments, 3-4 fractions containing the highest amount of prodrug and thus corresponding to liposome elution were pooled. The experiments on isolation of liposome–protein complexes were repeated at least twice for each type of liposomes.
Total protein was determined according to a modified Lowry procedure [17]. To prepare reagent A, 2 g Na2CO3, 0.2625 g potassium-sodium tartrate, 0.4 g NaOH, and 1 g SDS were dissolved in 100 ml bidistilled water. Reagent C was prepared immediately before use by the addition of one volume of 4% CuSO4 solution (reagent B) to 100 volumes of reagent A. Then, 300 µl of reagent C was added to a 100-µl analyzed sample, shaken, and left for 10 min. Then 30 µl Folin reagent diluted with distilled water (1 : 1) was added. After 60 min, optical density was measured at 660 nm. Control sample contained 100 µl PBS.
Delipidation of joint protein fractions and SDS-PAGE. Delipidation was performed as described in [18]. Four hundred microliters of cold MeOH was added to 100 µl pooled fractions, mixed, and centrifuged for 3 min at 9000g. Two hundred microliters of CHCl3 was added to the solution, actively mixed, and centrifuged for 3 min at 9000g. Then, 300 µl H2O was added to the mixture, actively shaken, and centrifuged for 4 min at 9000g; phase separation was observed with protein concentrating at the interphase. Seven hundred microliters of the upper phase was removed. Another 300 µl MeOH was added, mixed, and centrifuged for 4 min at 9000g. A small pellet was observed on the bottom of the tube. The supernatant was decanted, and the remaining ~30-50 µl liquid was evaporated on a rotary evaporator. Twenty microliters of 2× SDS gel-loading buffer (1.25 M Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 10% β-mercaptoethanol, and 0.02% bromophenol blue) was added to samples, mixed, and heated on a water bath (90-95°C) for 2 min twice with active shaking. Electrophoresis according to Laemmli [19] was performed in a 6% concentrating and 10-12.5% separating gel on a Helicon VE-2M (Helicon, Russia) apparatus at 200 V. Proteins were visualized by Coomassie G-250 or silver staining [20]. Electrophoregrams were analyzed with ImageJ software. To determine protein molecular weights, SigmaMarker (Sigma) or Prestained Protein Molecular Weight Marker (Fermentas, Lithuania) was used.
Immunoblotting. Proteins were transferred onto a Hybond-P Amersham (GE Healthcare, USA) PVDF membrane at 15 V during 40-60 min using a system for semi-dry transfer from Helicon. To prevent nonspecific adsorption, the membrane was incubated with 5% low-fat dry milk solution in 0.05 M Tris-buffered saline, pH 7.9, containing 0.1% Tween 20 for an hour at room temperature. The membrane was washed and incubated with primary antibodies (sheep polyclonal antibodies to human C4b-binding protein, ab8788; AbCam, USA; goat antibodies to C3 component, A213, and factor H, A237, of the human complement; ComplementTech, USA; rabbit polyclonal antibodies to human apolipoprotein AI, RAH Laa, and fibronectin, RAH Fne, and monoclonal mouse antibodies to human apolipoprotein E, MGH Lee; IMTEK, Russia) to plasma proteins in a 0.5% BSA solution overnight at 4°C. The membrane was washed, incubated with horseradish peroxidase-conjugated secondary IgG (rabbit antibodies to sheep and goat IgG; Jackson ImmunoResearch, USA; goat antibodies to rabbit and mouse IgG; Sigma), and again thoroughly washed. Immunodetection was performed with Immun-StarTM HRP Substrate (Bio-Rad, USA) reagent and a VersaDos 4000 (Bio-Rad) system. We are grateful to Dr. E. P. Kopantsev (Institute of Bioorganic Chemistry, Russian Academy of Sciences) for his help with immunodetection.
RESULTS AND DISCUSSION
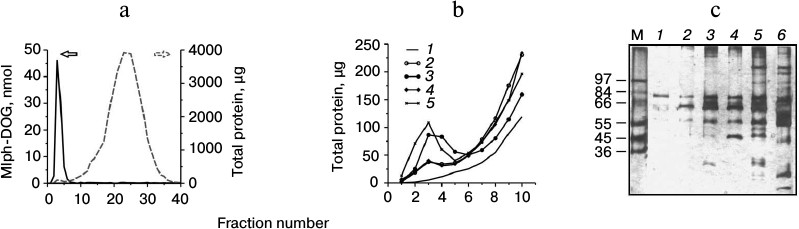
To study binding of blood plasma proteins, liposome preparations were incubated in 80% human plasma for 15 min, since results implying interaction between the studied liposomes and proteins of the complement and coagulation cascades were obtained upon short-term incubation [13]. Then, using gel-permeation chromatography on Sepharose, fractions containing liposomes were separated; total protein measured in the fractions was related to the total amount of lipids in them, thus producing the protein binding (PB) capacity value. Typical elution profiles are presented in Figs. 2a and 2b. In the case of MTX-DOG liposomes (MTX-L), the PB value was 83.9 ± 12.7 g protein/mol lipids, and for Mlph-DOG liposomes (Mlph-L) it was 72.6 ± 3.0 g protein/mol lipids (mean values for two independent experiments).
Fig. 2. a) Liposome and protein elution profiles during gel-permeation chromatography on Sepharose CL-4B after incubation of liposomes with 80% plasma for 15 min at 37°C (by the example of Mlph-L); the prodrug was identified by UV spectrophotometry, and protein was determined by a modified Lowry procedure. b) Comparison of protein elution profiles after liposome sample incubation in 80% plasma. c) Separation of liposome-associated proteins with SDS-PAGE according to Laemmli (aliquots contained equal amounts of lipids): 1) control (PBS); 2) liposomes prepared from mixture of PC–PI 8 : 1; 3) Mlph-L (10 mol% Mlph-DOG prodrug in the bilayer); 4) MTX-L-2.5 (2.5 mol% MTX-DOG prodrug in the bilayer); 5) MTX-L (10 mol% MTX-DOG prodrug in the bilayer); 6) plasma diluted 1 : 500; M) molecular mass markers.
The derived PB values allow for prognosis of rapid elimination of liposomes from circulation. For example, liposomes binding more than 50 g protein/mol lipids are characterized by plasma half-lives (T1/2) of less than 2 min [6]; the values are typical for anionic liposomes containing up to 20% of such lipids as phosphatidic acid, phosphatidylserine, and cardiolipin [4]. On the contrary, neutral liposomes, for example liposomes based on mixtures of phosphatidylcholine and cholesterol, binding less than 20 g protein per mol lipid, remain in circulation much longer, with T1/2 values over 2 h [6]. Intermediate PB values typical of negatively charged liposomes containing phosphatidylglycerol and phosphatidylinositol correspond to intermediate values of liposome circulation half-lives [6]. The studied liposomes are also negatively charged: ζ-potential values for MTX-L and Mlph-L are –53 ± 4 and –34 ± 3 mV, respectively [13]. Control liposomes built of matrix lipids (PC/PI) and with an intermediate surface charge (ζ-potential of –42 ± 1 mV) bound much less protein, PB = 26.8 ± 2.2 g protein per mol lipid. Similarly, liposomes containing less MTX-DOG in the bilayer (2.5 mol%) demonstrated lower PB value (32.8 ± 6.3 g protein per mol lipid) even with their considerable value of ζ-potential of –45 ± 2 mV. Therefore, in agreement with data in the literature [6], the interaction of liposomes with plasma protein is determined not only by the charge of the liposome surface, but also by its structure, which is mainly determined by the polar head groups of the lipid bilayer.
Interestingly, the PB value obtained for control liposomes is practically the same as the one obtained by other researchers for liposomes made of plant phosphatidylinositol (27 g protein/mol lipid), which remained in circulation for a rather long time, T1/2 = 90 min [6]. Phosphatidylinositol in liposomes is known to act as an anti-opsonizing factor, since the highly hydrophilic myoinositol residues promote steric stabilization of the surface similar to long chains of PEG [21, 22]. In contrast to PEGylated liposomes (for example, [23]), our control preparation did not activate the C system [13].
Analysis of protein binding profiles for different liposomes with standard denaturing gel electrophoresis revealed differences between the formulations (Fig. 2c). On the basis of the pattern of a nonspecifically stained gel, we may already assume some proteins involved in interactions with liposomes. As a rule, liposome–protein complexes contain major (by mass) proteins of plasma, that is albumin, immunoglobulin G, and fibrinogen, even if their affinity to the liposome surface is rather low [24] (with time, they are replaced by proteins less abundant in plasma but exhibiting higher affinity to the liposome surface; this is the so-called Vroman effect [25, 26]). Indeed, in all samples the band corresponding to a protein of 66 kDa is dominating. Most probably, it represents serum albumin (60% to the total plasma protein). A band corresponding to ~55 kDa is, most likely, fibrinogen monomer, another abundant plasma protein. Other pronounced bands were preliminarily referred to α-, α2-, and β-chains of complement factor C3 (~130, 45, and 73 kDa, respectively), heavy chain of IgG (~50 kDa), IgM (75 kDa), and C4b-binding protein (75 kDa α-subunit and 45 kDa β-subunit).
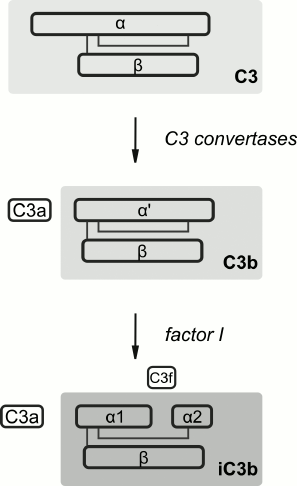
Specific detection of individual proteins associated with liposomes was performed with immunoblotting. Taking into account the data on C activation by liposomes with MTX-DOG [13], binding of proteins of the C system was our main interest. The C3 component is the most abundant among the C proteins (1.2 mg/ml plasma) and consists of two polypeptide chains, α- (116 kDa) and β-subunit (68 kDa; Fig. 3) [27]. Complement activation via three main pathways leads to hydrolysis of the α-subunit of C3 and release of anaphylatoxin C3a (detected in plasma in the presence of MTX-L with ELISA [13]) and the C3b fragment (α′ + β). The regulatory component of the alternative C activation pathway, factor H, competes with factors B and C5 for binding with C3b, including the surface-associated C3b, thus inhibiting amplification of the signal in the amplification loop of the alternative activation pathway [28]. Factor H also replaces fragment Bb in the active convertases C3 and C5. In the presence of factor H, factor I (fI) hydrolyzes the C3b fragment, releasing the 3-kDa fragment C3f and inactive iC3b (its fragments α1, α2, and β typically are detected by electrophoresis under reducing conditions). The iC3b fragment is not able to bind Bb to form C3 convertases; however, when binding the pathogen surface, it is an active opsonizing agent.
Fig. 3. Schematic structure and transformations of complement component C3 (adapted from [13]).
We observed C3 fragments – α- and β-subunits, as well as fragment α2 – only among the proteins associated with MTX-L (Fig. 4a). The band corresponding to the full α-subunit is weakly stained, evidencing hydrolysis of C3 under the effect of convertases formed upon C activation. It is reasonable to assume the following progression of events. Fragment C3b binds the MTX-L surface and is being cleaved under the effect of factor I in the presence of factor H, which was also detected in abundance in a complex with MTX-L (Fig. 4b). The iC3b fragment thus formed remains bound with the liposome surface. Importantly, neither fragment C3 nor factor H was detected among plasma proteins associated with liposomes with decreased MTX-DOG content (2.5 mol%, the MTX-L-2.5 sample). Most likely this is due to a different architecture of MTX-L-2.5 liposomes free of strains and deformations in the bilayer supposedly characteristic of MTX-L liposomes containing 10 mol% prodrug.
Fig. 4. Identification of liposome-associated proteins by immunoblotting using antibodies to the components of complement C3 (a), factor H (b), and C4b-binding protein (c). See Fig. 2 caption for sample designation.
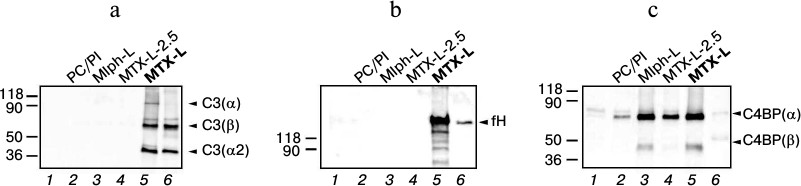
C4b-binding protein (C4BP) is a multimer protein synthesized in the liver. The most abundant form comprises seven α-subunits (75 kDa each) and a single β-subunit (45 kDa) [27]. C4BP promotes dissociation of convertases of the classical and lectin pathways of C activation and is a cofactor of fI in reaction of C4b factor hydrolysis. Since considerable binding of C4BP (Fig. 4c) was detected for both MTX-L inducing C activation in vitro, and Mlph-L not activating C, it is unlikely that this regulatory protein influences C functioning in the presence of the studied liposomes.
Interestingly, binding of factor H (fH) on surfaces normally serves for differentiation of host and pathogen cells by complement. Factor H is considered to bind the surface of cells, first of all endothelial cells in constant contact with plasma, through specific interactions with integrins, for example CD11b/CD18, as well as nonspecific ones with polyanionic structures such as sialic acids, heparin sulfates, and glycosaminoglycans ([29] and references therein). Therefore, fH participates in inhibition of C activation in close proximity to a cell surface. Binding of fH and adsorbed C3b requires an anionic surface [30]. In the case of MTX-L, two possible sequences of events can be proposed: 1) C activation in the presence of liposomes initiated via one of the three pathways leads to formation of a surface-associated C3b fragment, but amplification of the signal via the alternative pathway is inhibited by fH, which, however, does not affect the resulting formation of the lytic, or membrane attack, complex and, consequently, lysis of sensitized erythrocytes as observed in vitro [13]; 2) complement is activated, but fH bound to the liposome surface undergoes conformational changes leading to the loss of its ability to regulate the response.
Fibronectin (molecular mass of the monomer is 235 kDa) is one of the major plasma proteins (~300 µg/ml); it belongs to non-immune opsonins. Fibronectin has been reported to bind liposomes actively [4, 31]. In addition, it has been shown to promote internalization of liposomes by phagocytes [32]. We observed an only insignificant amount of fibronectin in the MTX-L-associated protein fractions (Fig. 5a, lane 5). Equally small amounts were registered in control (diluted) plasma, which is most probably explained by the insufficient sensitivity of the immunoblotting technique, which is typical of large polypeptides.
Fig. 5. Identification of liposome-associated proteins by immunoblotting using antibodies to fibronectin (a) and apolipoproteins AI (b) and E (c). See Fig. 2 caption for sample designation.

Apolipoprotein AI (ApoAI, 28.3 kDa) is part of high-density lipoproteins and chylomicrons and is a lecithin-cholesterol acyltransferase activator, thus providing for the reverse transport of cholesterol from peripheral tissues to liver. Similar to other apolipoproteins, ApoAI is able to affect biodistribution and pharmacokinetics of liposomes. ApoAI is known to participate in destabilization of lipid vesicles in the circulation [33]. Under the conditions of our experiment, ApoAI could not be detected in any of the liposome samples (Fig. 5b), which may be explained by the absence of cholesterol in the lipid bilayer [34]. Amphipathic helical domains of ApoAI are known to hardly penetrate fluid (with low phase-transition temperature) lipid bilayers built from phospholipids with non-saturated acyl chains [33]. Therefore, no concentrating of ApoAI occurs at the liposome surface (Fig. 5b, lane 1) and the protein is not likely to participate in liposome withdrawal from the circulation.
Apolipoprotein E (ApoE, 34 kDa) is a major component of chylomicrons and intermediate-density lipoproteins. Owing to its high affinity to the family of low-density lipoprotein (LDL) receptors, ApoE plays a key role in transport and metabolism of triglyceride-rich lipoprotein fractions. Binding of ApoE with all liposome samples (Fig. 5c, lanes 2-6) may evidence that in circulation liposomes will be directed to various organs and tissues expressing LDL receptors. However, according to a number of publications ([33] and references therein), neither ApoE nor LDL receptors play a significant role in catabolism of phosphatidylcholine liposomes.
The reason for reactivity of MTX-DOG liposomes may lie in both the deformation of the liposome surface organization by the bulky methotrexate moieties and the presence of two exocyclic aromatic amino groups and a free α-carboxyl group. For example, functionalization of the surface of phospholipid nanoparticles with lipid derivatives of gadolinium chelates resulted in C activation via the classical pathway (i.e. was initiated by IgG binding), and the intensity of the response depended not on ζ-potential of the particle surface, but rather on the chelate structure [35]. On the other hand, exposed amino (and hydroxyl) groups arranged in a specific manner may cause complement activation through nucleophilic attack on the internal thioester bond of the C3b fragment [36], leading to acceleration of the spontaneous hydrolysis of C3 and activation of the alternative pathway [37].
The mechanisms of C activation by liposomes of different compositions remain poorly studied. Earlier, negatively charged liposomes have been shown to activate C via the classical pathway, as well as through direct interactions of the C1q component with anionic lipids ([38] and references therein). On the contrary, positively charged liposomes activate C via the alternative pathway [39]. The studied liposomes are negatively charged, and at the same time only formulations with high MTX-DOG content in the bilayer (10 mol%) that induced C activation [13] bound fragments of C3 and fH, regulating C activation via the alternative pathway. Probably, aromatic NH2 groups of the MTX-DOG molecules arranged in a special manner on the bilayer surface may indeed react with the internal thioester bond in the C3b fragment and thus accelerate spontaneous hydrolysis of the C3 component, leading to activation of the alternative pathway (see above). Independently of the activation mechanism, the observed differences in protein-binding profiles are in agreement with the data on complement activation in in vitro tests. Indeed, upon decrease in the liposome load with the prodrug down to 2.5 mol% (MTX-L-2.5 samples; corresponds to methotrexate low-dose treatment regimen), no binding of C components was observed and C activation proceeded at an insignificant level [13]. Therefore, the liposome composition defines the surface properties, which in turn determines the set of plasma proteins bound and thus causes inertness or reactivity of liposomes in circulation. The pathways of C activation realized upon contact with liposomes and the manifestations of these interactions between liposomes and blood plasma proteins in vivo are subjects of our ongoing studies.
This work was supported by the Russian Foundation for Basic Research (project No. 12-04-31739mol_a).
REFERENCES
1.Monopoli, M. P., Aberg, C., Salvati, A., and
Dawson, K. A. (2012) Biomolecular coronas provide the biological
identity of nanosized materials, Nat. Nanotech., 7,
779-786.
2.Walkey, C. D., and Chan, W. C. W. (2012)
Understanding and controlling the interaction of nanomaterials with
proteins in a physiological environment, Chem. Soc. Rev.,
41, 2780-2799.
3.Aggarwal, P., Hall, J. B., McLeland, C. B.,
Dobrovolskaia, M. A., and McNeil, S. E. (2009) Nanoparticle interaction
with plasma proteins as it relates to particle biodistribution,
biocompatibility and therapeutic efficacy, Adv. Drug Deliv.
Rev., 61, 428-437.
4.Semple, S. C., Chonn, A., and Cullis, P. R. (1998)
Interactions of liposomes and lipid-based carrier systems with blood
proteins: relation to clearance behavior in vivo, Adv. Drug
Deliv. Rev., 32, 3-17.
5.Dobrovolskaia, M. A., Neun, B. W., Man, S., Ye, X.,
Hansen, M., Patri, A. K., Crist, R. M., and McNeil, S. E. (2014)
Protein corona composition does not accurately predict
hematocompatibility of colloidal gold nanoparticles,
Nanomedicine, doi: 10.1016/j.nano.2014.01.009.
6.Chonn, A., Semple, S. C., and Cullis, P. R. (1992)
Association of blood proteins with large unilamellar liposomes in
vivo. Relation to circulation lifetimes, J. Biol. Chem.,
267, 18759-18765.
7.Szebeni, J. (2012) Hemocompatibility testing for
nanomedicines and biological: predictive assays for complement mediated
infusion reactions, Eur. J. Nanomed., 4, 33-53.
8.Szebeni, J., Muggia, F., Gabizon, G., and
Barenholz, Y. (2011) Activation of complement by therapeutic liposomes
and other lipid excipient-based therapeutic products: prediction and
prevention, Adv. Drug Deliv. Rev., 63, 1020-1030.
9.Szebeni, J., Muggia, F. M., and Alving, C. R.
(1998) Complement activation by Cremophor EL as a possible contributor
to hypersensitivity to paclitaxel: an in vitro study, J.
Natl. Cancer Inst., 90, 300-306.
10.Kuznetsova, N., Kandyba, A., Vostrov, I.,
Kadykov, V., Gaenko, G., Molotkovsky, J., and Vodovozova, E. (2009)
Liposomes loaded with lipophilic prodrugs of methotrexate and melphalan
as convenient drug delivery vehicles, J. Drug Deliv. Sci.
Technol., 19, 51-59.
11.Vodovozova, E. L., Moiseeva, E. V., Grechko, G.
K., Gayenko, G. P., Nifant’ev, N. E., Bovin, N. V., and
Molotkovsky, J. G. (2000) Antitumor activity of cytotoxic liposomes
equipped with selectin ligand SiaLeX, in a mouse mammary adenocarcinoma
model, Eur. J. Cancer, 36, 942-949.
12.Kozlov, A. M., Korchagina, E. Yu., Vodovozova, E.
L., Bovin, N. V., Molotkovskii, Yu. G., and Syrkin, A. B. (1997)
Increase in sarcolysin antitumor activity by transforming it into a
lipid derivative and incorporation into the membrane of liposomes
containing a carbohydrate vector, Byul. Eksp. Biol. Med.,
123, 381-383.
13.Kuznetsova, N. R., Sevrin, C., Lespineux, D.,
Bovin, N. V., Vodovozova, E. L., Meszaros, T., Szebeni, J., and
Grandfils, C. (2012) Hemocompatibility of liposomes loaded with
lipophilic prodrugs of methotrexate and melphalan in the lipid
bilayer, J. Control Rel., 160, 394-400.
14.Heiss, H. W. (1983) in Human Physiology
(Schmidt, R. F., and Thews, G., eds.) Springer-Verlag,
Berlin-Heidelberg-New York, pp. A43-A44.
15.Vodovozova, E. L., Nikol’skii, P. Yu.,
Mikhalev, I. I., and Molotkovskii, Yu. G. (1996) Lipid derivatives of
sarcolysin, methotrexate, and rubomycin, Russ. J. Bioorg.
Chem., 22, 468-475.
16.Vodovozova, E. L., Gaenko, G. P., Bobrikova, E.
S., Pazynina, G. V., and Molotkovskii, Yu. G. (2007) A diglyceride
derivative of methotrexate: synthesis and cytotoxic activity in
targeted delivery liposomes, Pharm. Chem. J., 41,
297-301.
17.Markwell, M., Haas, S., and Bieber, L. (1978) A
modification of the Lowry procedure to simplify protein determination
in membrane and lipoprotein samples, Anal. Biochem., 210,
206-210.
18.Dos Santos, N., Allen, C., Doppen, A.-M.,
Anantha, M., Cox, K., Gallagher, R. C., Karlsson, G., Edwards, K.,
Kenner, G., Samuels, L., Webb, M. S., and Bally, M. B. (2007) Influence
of poly(ethylene glycol) grafting density and polymer length on
liposomes: relating plasma circulation lifetimes to protein binding,
Biochim. Biophys. Acta, 1768, 1367-1377.
19.Laemmli, U. (1970) Cleavage of structural
proteins during the assembly of the head of bacteriophage T4,
Nature, 227, 680-685.
20.Shevchenko, A., Wilm, M., Vorm, O., and Mann, M.
(1996) Mass spectrometric sequencing of proteins silver-stained
polyacrylamide gels, Anal. Chem., 68, 850-858.
21.Gabizon, A., and Papahadjopoulos, D. (1988)
Liposome formulations with prolonged circulation time in blood and
enhanced uptake by tumors, Proc. Natl. Acad. Sci. USA,
85, 6949-6953.
22.Peng, A., Straubinger, R. M., and Balu-Iyer, S.
V. (2010) Phosphatidylinositol containing lipidic particles reduces
immunogenicity and catabolism of factor VIII in hemophilia a mice,
AAPS J., 12, 473-481.
23.Moghimi, S. M., Andersen, A. J., Hashemi, S. H.,
Lettiero, B., Ahmadvand, D., Hunter, A. C., Andresen, T. L., Hamad, I.,
and Szebeni, J. (2010) Complement activation cascade triggered by
PEG-PL engineered nanomedicines and carbon nanotubes: the challenges
ahead, J. Control Rel., 146, 175-181.
24.Cedervall, T., Lynch, I., Lindman, S., Berggard,
T., Thulin, E., Nilsson, H., Dawson, K. A., and Linse, S. (2007)
Understanding the nanoparticle-protein corona using methods to quantify
exchange rates and affinities of proteins for nanoparticles, Proc.
Natl. Acad. Sci. USA, 104, 2050-2055.
25.Vroman, L., Adams, A., Fischer, G., and Munoz, P.
(1980) Interaction of high molecular weight kininogen, factor XII, and
fibrinogen in plasma at interfaces, Blood, 55,
156-159.
26.Goppert, T. M., and Muller, R. H. (2005)
Adsorption kinetics of plasma proteins on solid lipid nanoparticles for
drug targeting, Int. J. Pharm., 302, 172-186.
27.Sahu, A., and Lambris, J. D. (2001) Structure and
biology of complement protein C3, a connecting link between innate and
acquired immunity, Immunol. Rev., 180, 35-48.
28.Soames, C. J., and Sim, R. B. (1997) Interactions
between human complement components factor H, factor I and C3b,
Biochem. J., 326, 553-561.
29.Jozsi, M., Manuelian, T., Heinen, S., Oppermann,
M., and Zipfel, P. F. (2004) Attachment of the soluble complement
regulator factor H to cell and tissue surfaces: relevance for
pathology, Histol. Histopathol., 19, 251-258.
30.Rodriguez de Cordoba, S., Esparza-Gordillo, J.,
Goicoechea de Jorge, E., Lopez-Trascasa, M., and Sanchez-Corral, P.
(2004) The human complement factor H: functional roles, genetic
variations and disease associations, Mol. Immunol., 41,
355-367.
31.Price, M. E., Cornelius, R. M., and Brash, J. L.
(2001) Protein adsorption to polyethylene glycol modified liposomes
from fibrinogen solution and from plasma, Biochim. Biophys.
Acta, 1512, 191-205.
32.Aramaki, Y., Akiyama, K., Hara, T., and Tsuchiya,
S. (1995) Recognition of charged liposomes by rat peritoneal and
splenic macrophages: effects of fibronectin on the uptake of charged
liposomes, Eur. J. Pharm. Sci., 3, 63-70.
33.Rodrigueza, W. V., Phillips, M. C., and Williams,
K. J. (1998) Structural and metabolic consequences of
liposome–lipoprotein interactions, Adv. Drug Deliv. Rev.,
32, 31-43.
34.Saito, Y. M., Handa, T., and Miyajima, K. (1997)
Effect of cholesterol on apolipoprotein A-I binding to lipid bilayers
and emulsions, J. Lipid Res., 38, 287-294.
35.Pham, C. T. N., Mitchell, L. M., Huang, J. L.,
Lubniewski, C. M., Schall, O. F., Killgore, J. K., Pan, D., Wickline,
S. A., Lanza, G. M., and Hourcade, D. E. (2011) Variable
antibody-dependent activation of complement by functionalized
phospholipid nanoparticle surfaces, J. Biol. Chem., 286,
123-130.
36.Janssen, B. J. C., Christodoulidou, A., McCarthy,
A., Lambris, J. D., and Gros, P. (2006) Structure of C3b reveals
conformational changes that underlie complement activity,
Nature, 444, 213-216.
37.Moghimi, S. M., Andersen, A. J., Ahmadvand, D.,
Wibroe, P. P., Andresen, T. L., and Hunter, C. (2011) Material
properties in complement activation, Adv. Drug Deliv. Rev.,
63, 1000-1007.
38.Devine, D. V., and Marjan, J. M. (1997) The role
of immunoproteins in the survival of liposomes in the circulation,
Crit. Rev. Ther. Drug Carrier Syst., 14, 105-131.
39.Devine, D. V., Wong, K., Serrano, K., Chonn, A.,
and Cullis, P. R. (1994) Liposome-complement interactions in rat serum:
implications for liposome survival studies, Biochim. Biophys.
Acta, 1191, 43-51.