Synthesis in Escherichia coli and Characterization of Human Recombinant Erythropoietin with Additional Heparin-Binding Domain
A. S. Karyagina1,2,3,a*, T. M. Grunina1, M. S. Poponova1, P. A. Orlova1, V. N. Manskikh1,3, A. V. Demidenko1, N. V. Strukova1, M. S. Manukhina1, K. E. Nikitin1, A. M. Lyaschuk1, Z. M. Galushkina1, S. A. Cherepushkin4, N. B. Polyakov1,5, A. I. Solovyev1, V. G. Zhukhovitsky1, D. A. Tretyak6, I. S. Boksha1,7, A. V. Gromov1,b*, and V. G. Lunin1,2
1Gamaleya National Research Center of Epidemiology and Microbiology, Ministry of Health of the Russian Federation, 123098 Moscow, Russia2All-Russia Research Institute of Agricultural Biotechnology, 127550 Moscow, Russia
3Belozersky Institute of Physical and Chemical Biology, Lomonosov Moscow State University, 119991 Moscow, Russia
4State Research Institute of Genetics and Selection of Industrial Microorganisms, Kurchatov Institute National Research Centre, 117545 Moscow, Russia
5Vernadsky Institute of Geochemistry and Analytical Chemistry, Russian Academy of Sciences, 119334 Moscow, Russia
6Moscow Technological University (Lomonosov Institute of Fine Chemical Technologies), 119571 Moscow, Russia
7Research Center of Mental Health, 115522 Moscow, Russia
* To whom correspondence should be addressed.
Received May 18, 2018
Recombinant human erythropoietin (EPO) with additional N-terminal heparin-binding protein domain (HBD) from bone morphogenetic protein 2 was synthesized in Escherichia coli cells. A procedure for HBD-EPO purification and refolding was developed for obtaining highly-purified HBD-EPO. The structure of recombinant HBD-EPO was close to that of the native EPO protein. HBD-EPO contained two disulfide bonds, as shown by MALDI-TOF mass spectrometry. The protein demonstrated in vitro biological activity in the proliferation of human erythroleukemia TF-1 cell test and in vivo activity in animal models. HBD-EPO increased the number of reticulocytes in the blood after subcutaneous injection and displayed local angiogenic activity after subcutaneous implantation of demineralized bone matrix (DBM) discs with immobilized HBD-EPO. We developed a quantitative sandwich ELISA method for measuring HBD-EPO concentration in solution using rabbit polyclonal serum and commercial monoclonal anti-EPO antibodies. Pharmacokinetic properties of HBD-EPO were typical for bacterially produced EPO. Under physiological conditions, HBD-EPO can reversibly bind to DBM, which is often used as an osteoplastic material for treatment of bone pathologies. The data on HBD-EPO binding to DBM and local angiogenic activity of this protein give hope for successful application of HBD-EPO immobilized on DBM in experiments on bone regeneration.
KEY WORDS: erythropoietin, Escherichia coli, heparin-binding domainDOI: 10.1134/S0006297918100061
Abbreviations: BCA, 2,2′-bicinchoninic acid; BMP-2, bone morphogenetic protein 2; DBM, demineralized bone matrix; DTT, dithiothreitol; EPO, erythropoietin; HBD, heparin-binding domain; 6His, six-histidine tag; s-tag, 15-amino acid oligopeptide from bovine pancreas ribonuclease A.
Erythropoietin (EPO) is a hormone (cytokine) that initiates
differentiation of erythrocyte precursors in bone marrow [1]. Apart from its role in erythropoiesis, EPO is
involved in wound healing, neuroregeneration, and other biological
processes, such as bone tissue remodeling that requires both angiogenic
and osteogenic activities of EPO [2-4]. The effect of recombinant eukaryotic EPO on bone
tissue regeneration in mammals has been demonstrated in various
experimental models [5-12], in
which EPO has been administered systemically or locally. However, there
are reasons to believe that local administration of EPO as a component
of implanted material is preferable in the treatment of bone
pathologies. Using the model of bone tissue regeneration in rabbit long
bones, Omlor et al. [12] demonstrated more than
two-fold increase in the volume of newly formed bone tissue at three
different time points (up to 12 weeks) after surgery upon local EPO
administration, as compared to a single systemic parenteral
introduction of the same amount of EPO. Multiple systemic parenteral
introduction of large doses of EPO may result in impaired hemostasis
(increased hematocrit and blood viscosity) and become a risk factor in
patients [13]. Therefore, high therapeutic
efficacy of EPO administered locally as a component of implanted
material makes EPO a promising alternative agent in the treatment of
bone pathologies.
Recombinant human EPO used in all the above cited works, as well as in clinical studies, was synthesized in cultured eukaryotic cells (Chinese hamster ovary cells) [14]. Produced molecular forms of EPO have identical amino acid sequence (165 a.a.) but differ in glycosylation and pharmacokinetic profiles: epoetin alpha, epoetin beta, darbepoetin alpha (hyperglycosylated analog of human recombinant epoetin alpha with the elimination half-life after intravenous administration about three times longer than of epoetin alpha and native EPO) [15]. The use of different EPO forms explains the differences in the treatment efficacy and results of model experiments. Besides, common problems in the synthesis of glycosylated recombinant proteins in eukaryotic systems are low yield and synthesis of proteins with different glycosylation profiles requiring their further fractionation.
An alternative approach to the biosynthesis of human recombinant EPO is heterologous prokaryotic expression system in Escherichia coli cells [16-20]. Using this system, EPO could be produced in high amounts in a more standardized state and at much less expense. However, bacterially produced EPO is not glycosylated, which makes it low soluble and prone to aggregation. Compared to the glycosylated forms, non-glycosylated proteins have lower molecular masses favoring their more rapid elimination from the circulation. The problem of low solubility can be solved by introducing amino acid substitutions at the sites of glycosylation [19] or by modifying EPO with additional protein domains that could be later cleaved off by proteolytic hydrolysis [17, 20]. The molecular mass of recombinant protein can be increased by various methods including the widely used procedure of PEGylation [16]. Since the synthesis of eukaryotic proteins in bacterial cells can be accompanied by formation of random disulfide bonds, formation of two correct S–S bonds typical for the native EPO requires additional refolding stage. The protein yield after purification is determined by the portion of correctly refolded molecules [20]. Nevertheless, recombinant EPO produced in E. coli cells displays biological activity [16-20]; hence, development of new procedures for recombinant EPO application, in particular, for regeneration of damaged bone tissue, is a promising approach in clinical practice.
In order to the recombinant EPO exert a long-term action after introduction to the bone defect, its release from the implanted material should be very slow. When using glycosaminoglycan-containing demineralized bone matrix (DBM) as a carrier, the recombinant protein could be retained in the matrix if it contains heparin-binding domain (HBD), an amino acid sequence that forms complexes with heparin and other negatively charged polysaccharides via ionic interactions. HBDs are present in many proteins, e.g., bone morphogenetic protein 2 (BMP-2). Due to the presence of HBD, BMP-2 can be purified on heparin-Sepharose [21] or immobilized on DBM [22]. We suggested that introduction of HBD into the molecule of recombinant EPO will promote EPO retention at the site of implantation if DBM is used as a carrier.
The goal of this work was to produce highly purified HBD-EPO preparations with correctly formed disulfide bonds. The recombinant HBD-EPO should be capable of binding with DBM and display in vitro and in vivo activities, so that it can be used in experiments on regeneration of bone tissue defects.
MATERIALS AND METHODS
Escherichia coli M15 [pREP4] (F– Φ80ΔlacM15, thi, lac–, mtl–, recA+, KmR) strain and plasmid vector pQE6 (Qiagen, USA) were used.
HBD-EPO cloning. The synthetic gene encoding HBD from Danio rerio BMP-2 fused to human EPO and flanked with the NcoI and Kpn2I restriction endonuclease sites at the 5′- and 3′-ends, respectively, was synthesized by Eurogen (Russia). Codon composition of the HBD-EPO gene was optimized using the JCat software (http://www.jcat.de/); the secondary structure of the transcribed RNA was corrected using the DINAMelt web server (http://mfold.rna.albany.edu/?q=DINAMelt/Two-state-folding). The synthesized gene was cloned into pQE6 vector by the NcoI and Kpn2I sites to generate the pL610 plasmid that was used to transform E. coli M-15 [pREP4] cells.
Calculated molecular mass and other properties of HBD-EPO. The characteristics of HBD-EPO were theoretically calculated using the https://web.expasy.org/protparam/ resource. The calculated molecular mass of HBD-EPO is 20520.51 Da; theoretical isoelectric point (pI) is 9.62. The absorbance of HBD-EPO solution in water (1 mg/ml) at 280 nm is 1.107 (protein with two disulfide bonds) or 1.095 (reduced protein).
Cultivation of transformed E. coli cells. Transformed E. coli cells were grown in LB medium containing appropriate antibiotics on a shaker at 180 rpm at 37°C to OD600 of 1.0-1.2. Protein synthesis was induced with 0.5 mM IPTG, and the cells were incubated for another 6 h at 180 rpm at 30°C. The cells were collected by centrifugation for 30 min at 5000g at 10°C and stored at –20°C.
Disintegration of E. coli cells and isolation of inclusion bodies. Frozen bacterial biomass (1 g) was thawed and suspended at a 1 : 10 ratio (w/v) in the lysing buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF)) containing 100 μg/ml lysozyme. The cell suspension was incubated for 20 min at room temperature, and the cells were disintegrated on ice by sonication with a Sonics Vibracell sonicator (Sonics, USA) (40% amplitude; 2.5 min; 3-s pulses with 2-s intervals). The resulting homogenate was centrifuged for 30 min at 20,000g at 10°C. The pellet containing inclusion bodies was washed twice with the lysing buffer and once with 20 mM Tris-HCl (pH 8.0).
Column chromatography and electrophoresis. Column chromatography on WorkBeads 40 S (Bio-Works, Sweden) and heparin-Sepharose CL-6B (Amersham Pharmacia Biotech, Sweden) was performed with Äkta Start chromatography system (GE Healthcare Life Sciences, USA). Protein concentration was determined from the protein solution absorbance at 280 nm using calculated extinction coefficients and with BCA Protein Assay (AppliChem, Germany).
Preparation of protein samples, electrophoresis in polyacrylamide gel (PAGE), gel staining, and gel densitometry were performed as described by Grunina et al. [20]. Protein molecular weight markers (14-97 kDa) were from Bio-Rad (USA).
Chromatography on WorkBeads 40 S. The washed inclusion bodies (0.25 g) were dissolved in 8 M urea in 20 mM Tris-HCl (pH 8.0) at a 1 : 10 (w/v) ratio. The solution was incubated for 3 h and then centrifuged at 9000g for 30 min. The supernatant was loaded at a flow rate of 1 ml/min on a column with 10 ml of WorkBeads 40 S equilibrated with 8 M urea in 20 mM Tris-HCl (pH 8.0). The column was washed at 2 ml/min to remove non-bound proteins, and HBD-EPO was eluted at the same flow rate with 0-1.0 M linear gradient of NaCl concentration in the same buffer (total volume, 100 ml). Fractions (5 ml) with the maximal absorbance at 280 nm were combined and analyzed by electrophoresis. Protein concentration in the collected fractions was estimated from the absorbance at 280 nm and by the BCA method (both methods produced convergent results).
HBD-EPO refolding. Protein solution after chromatography on WorkBeads 40 S was diluted with 20 mM Tris-HCl (pH 8.0) containing 6 M guanidine hydrochloride and 0.1 M dithiothreitol (DTT) to a final protein concentration of ~0.1 mg/ml and incubated for 24 h at 4°C. The denatured protein was dialyzed against 10 volumes of 20 mM Tris-HCl (pH 8.0) containing 1 M guanidine hydrochloride for 24 h with three buffer changes and centrifuged for 30 min at 9000g. The supernatant was diluted with the refolding buffer (20 mM Tris-HCl, pH 8.5, containing 1 M L-arginine) at a 1 : 1 ratio to the final protein concentration of ~0.05 mg/ml and incubated for 24 h at 4°C. The refolded protein was dialyzed against 10 volumes of 20 mM Tris-HCl (pH 6.8) containing 4 M urea for 24 h and then centrifuged for 30 min at 9000g.
Affinity chromatography on heparin-Sepharose CL-6B. Protein solution after refolding was loaded at a flow rate of 1 ml/min on a column with 10 ml of heparin-Sepharose CL-6B equilibrated with 4 M urea in 20 mM Tris-HCl (pH 6.8). The column was washed with the same buffer, and the protein was eluted with 0-1.0 M linear gradient of NaCl concentration in the same buffer at the same flow rate. Four fractions (6.5 ml each) were collected; the first three fractions contained monomeric HBD-EPO, while the last fraction contained protein aggregates (seen in PAGE). Fractions containing monomeric HBD-EPO were combined (total volume, 19.5 ml) and dialyzed against 10 volumes of 20 mM ammonium acetate (pH 4.5) for 24 h at 4°C with three buffer changes. For dialysis, we used 25 μm-thick Zellu Trans/ROTH 3,5 E657.1 cellulose dialysis membrane with a 3500-Da cut-off. Protein concentration and total protein amount after dialysis was estimated from the absorbance at 280 nm and by the BCA method (both methods produced convergent results). The absorbance of the combined fraction (20 ml) was 0.181 OU, which corresponded to the protein concentration of 0.165 mg/ml (taking into account theoretical extinction coefficient for the oxidized protein). Purified protein preparations were lyophilized and used in further studies.
HBD-EPO hydrolysis with trypsin for mass-spectrometry. Proteins fractionated by PAGE were digested in the gel as described by Shevchenko et al. [23] in the presence or absence of DTT and iodoacetamide [20].
MALDI mass spectrometry. Peptides were analyzed with a time-of-flight MALDI Ultraflextreme mass spectrometer (Bruker Daltonics, Germany) using dihydroxybenzoic acid as a matrix. Positively charged ions were detected in the reflectron mode at 20.12 kV on the ion source IS1, 17.82 kV – on IS2, 7.47 kV – on lenses, 21.07 kV – on reflectron Ref1, and 10.80 kV – on Ref2.
The ions were detected within the m/z range of 700-5000 Th. Peaks corresponding to trypsin and keratin autolytic fragments were used as internal standards and excluded from the list of detected masses.
The spectra were analyzed with the Flex Analysis 3.4 software (Bruker Daltonics) and Proteinscape 4.1 program suit (Bruker Daltonics) using the following parameters: accuracy – 100 ppm; possible posttranslational modifications – oxidation of methionine, histidine, and tryptophan residues; cysteine modification with acrylamide. When analyzing the primary sequence, we also considered the possibility of cysteine modification with iodoacetamide. Identification of peptides permitted no more than six nonhydrolyzed trypsin sites per protein molecule.
In vitro biological activity assay. Biological activity of EPO preparations was assayed in the in vitro proliferation test in human erythroleukemia cell line TF-1 (ATCC CRL-2003) as suggested by Grunina et al. [20].
Biological activity of BMP-2 preparations was estimated in vitro from the increase in the activity of alkaline phosphatase synthesized by mouse C2C12 myoblasts in response to protein addition as described in [24, 25].
Estimation of HBD-EPO biological activity in vivo. Biological activity of EPO preparations was assayed in CBA mice [26] at the Olfarm Ltd. (Russia) laboratory. Normocythemic females (8 week-old) were injected subcutaneously with 0.5 ml of an equimolar mixture of heparin (molar mass, 8000-25,000 g/mol; BioChemica AppliChem, Germany) with tested EPO preparation at a concentration of 360, 180, or 90 μg/ml. Control mice were injected with physiological saline or Epostim (recombinant epoetin beta produced in eukaryotic cells; 100,000 IU/mg; Farmapark Ltd., Russia) in a concentration of 80, 40, or 20 IU/ml. Blood was collected 96 h after injection and analyzed for the reticulocyte count. Cells with a characteristic reticular network were counted in blood smears under a light microscope after supravital staining of nucleic acids in cell nuclei. The activity of HBD-EPO was estimated relatively to the control using the calibration curve.
Polyclonal anti-EPO serum. Polyclonal anti-EPO serum was obtained by immunization of chinchilla rabbits (body weight of 2.5-3.0 kg) with purified 6His-MBP-EPO and s-tag-EPO proteins [20] by the following scheme: first session – 5 injections 200 μl each (total of 150 μg 6His-MBP-EPO in physiological solution (1 : 1); complete Freund adjuvant) in the top of the shoulder and four zones of axillary and inguinal lymphatic nodes; 2-week break; second session – 5 injections 200 μl each (total of 150 μg 6His-MBP-EPO in physiological solution (1 : 1); incomplete Freund adjuvant) in the top of the shoulder and four zones of axillary and inguinal lymphatic nodes; 2-week break; third session – injection of 1 ml (150 μg) of s-tag-EPO in physiological solution in the thigh muscle; 1-week break; fourth session – injection of 1 ml (150 μg) of s-tag-EPO in physiological solution in another thigh muscle; 1-week break. The serum titer was determined by ELISA using HBD-EPO, s-tag-EPO, and Epostim in a concentration of 5 μg/ml as antigens and was estimated as no less than 1 : 2500 for each protein.
HBD-EPO immunoassay. Six different clones of commercially available monoclonal antibodies (Bialexa LTD, Russia) were tested for their interaction with HBD-EPO and Epostim by immunoblotting according to a standard procedure recommended by Amersham GE Healthcare. After electrophoresis, Epostim and HBD-EPO were transferred onto Hybond-C Extra nitrocellulose membrane using Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad, USA) at 2 mA/cm2; the membrane was blocked with 0.5% casein, cut into strips, and incubated for 1 h with EP1, EP2, EP3, EP5, EP8, and EP25 antibodies (Bialexa LTD) at a 1 : 1000 dilution and then for 1 h with rabbit anti-mouse immunoglobulins conjugated with horseradish peroxidase (1 : 10,000 dilution; IMTEK, Russia). The membranes were stained by the peroxidase reaction using hydrogen peroxide and diaminobenzidine.
Erythropoietin-EIA-Best kit for sandwich ELISA (Vektor-Best-Evropa, Russia) was used as recommended by the manufacturer, except that EPO concentration was determined in phosphate buffer instead of patient’s serum.
Sandwich ELISA for HBD-EPO quantitative assay in solution. The concentrations of HBD-EPO and Epostim were determined by indirect sandwich ELISA using EP8 monoclonal anti-EPO antibody (Bialexa LTD) as a capturing agent, rabbit anti-EPO serum as a detecting agent, and 1 mg/ml bovine serum albumin in phosphate buffer containing 0.1% Tween 20 as a blocking agent.
Pharmacokinetic studies were performed as described by Wang et al. [16] with slight modifications in six Wistar female rats (body weight of 270-280 g). The rats were divided into two groups (3 animals per group). Group 1 was injected with HBD-EPO; group 2 was injected with Epostim. The rats were injected subcutaneously in the top of the shoulder with equimolar mixture of a protein with heparin (molar mass, 8000-25,000 g/mol; BioChemica AppliChem, Germany) at a dose of 100 μg/kg body weight. Blood (200 μl) was collected from the tail vein into tubes with EDTA K3 0.5, 2, 5, 10, 24, 48, and 72 h after injection. After centrifugation at 400g for 15 min at 4°C, the plasma was collected and centrifuged at 2000g at room temperature for 5 min. The plasma was stored at –70°C, EPO concentration was determined using the developed ELISA test.
HBD-EPO binding to DBM. To obtain DBM membranes, diaphyses of bovine femoral bones were cut with band saw into 1- to 3-mm thick slices, that were then delipidated, decalcified, and deproteinized to remove non-collagen protein as described in [27] with modifications [22]. Disks (4 mm in diameter) were cut out, weighed, and standardized according to their mass. Each experiment used disks with maximally similar masses that depended on the amount of the applied protein.
To immobilize HBD-EPO, each disk was placed in 100 μl solution containing 1 to 10 μg HBD-EPO in 25 mM phosphate buffer (pH 5.5) and incubated with shaking for 3 h. The disks were then washed 3 times for 20 min with 1 ml of the same buffer and lyophilized. The amount of bound HBD-EPO was calculated from the content of HBD-EPO in the washing solution (i.e., not bound to the disk) measured by ELISA. To determine the amount of bound protein directly, each disk was incubated in 1 ml of 6 M guanidine hydrochloride at room temperature overnight on a shaker, and then the resulting wash out was dialyzed against 100 volumes of 25 mM phosphate buffer (pH 5.5) for 24 h. The amount of HBD-EPO was determined by the developed ELISA method. We found that under the conditions used, virtually all HBD-EPO bound to the DBM. After this, the disks were frozen, lyophilized, and sterilized by irradiation (adsorbed dose, 20 ± 5 kGy). For simultaneous immobilization of HBD-EPO and BMP-2 of DBM disks, the incubation solution contained 1 to 20 μg s-tag-BMP-2 [25] in addition to HBD-EPO. The amount of bound s-tag-BMP-2 was determined using the earlier developed ELISA test system [28].
The quality of the bound proteins and the ratio between them were estimated by PAGE. DBM disks with immobilized proteins were incubated with 50 μl Laemmli’s sample buffer containing 0.2 M DTT, 2 mM EDTA, 125 mM Tris-HCl (pH 6.8), 20% glycerol, 4% SDS, and 0.015% Bromophenol Blue for 5 min at 95°C and then centrifuged for 5 min at 16,000g. The supernatant was fractionated by PAGE.
HBD-EPO angiogenic activity. The angiogenic activity of HBD-EPO was assayed in six Wistar female rats (body weight of 270-280 g). The rats were divided into two groups (3 animals per group). The animals were anesthetized intravenously with Zoletil 100 (15 mg/kg body weight) and intraperitoneally with Rometar (5 mg/kg body weight). In each rat, two DBM disks with the same immobilized protein were implanted subcutaneously through 1-cm cuts in the temples symmetrically on both sides of the head. The first group was implanted with disks loaded with 1 μg HBD-EPO; the second group (control) was implanted with disks with immobilized 1 μg Epostim. To immobilize HBD-EPO and Epostim, the disks were placed in 1.5-ml plastic tubes containing 1 μg of protein in 20 μl of 25 mM phosphate buffer (pH 5.5), incubated overnight at 10°C, and used for implantation without further lyophilization.
The animals were euthanized with carbon dioxide 5 days later, and the disks with surrounding tissues were removed. One of the two disks from the same animal was fixed for 24 h in freshly prepared 10% formalin buffered solution (pH 7.4) and used for preparation of paraffin slides for hematoxylin-eosin and Heidenhain’s AZAN trichrome staining [29] with further histological analysis and histomorphometry. The second disk was used for determining the residual protein. For this, the disk was incubated in 1 ml of 6 M guanidine hydrochloride in 25 mM phosphate buffer at 4°C overnight on a shaker and then dialyzed against 100 volumes of 25 mM phosphate buffer (pH 5.5) at 4°C for 24 h. The amount of the residual protein was determined using the developed ELISA test system.
RESULTS AND DISCUSSION
To create EPO variants capable of retention on DBM, we needed to find small protein domain that could be introduced into EPO molecule to provide immobilization of the resulting chimeric protein on DBM. The most obvious candidate was heparin-binding domain (HBD) of BMP-2. The function of this domain is a long-term retention of BMP-2 in the bone tissue. HBD is located in the N-terminal part of BMP-2 molecule and contains a large number of positively charged amino acid residues. Since we planned to use the created recombinant protein in experiments on the bone tissue regeneration, in particular, in combination with BMP-2, it was undesirable to fuse EPO with amino acid sequence identical to the fragment of human BMP-2 because it could hinder its immunological detection in experiments on the binding and kinetics of EPO elution from DBM. We compared BMP-2 sequences from various vertebrate species (data not shown) and chose HBD from the zebra fish Danio rerio that is maximally distant from the human HBD (Fig. 1).
Fig. 1. Amino acid sequence of HBD-EPO. EPO sequence is shown in bold; HBD (D. rerio) is shown in italic; linker is underlined; cysteine residues in EPO are shown in bold and underlined. Amino acid sequence of HBD from Homo sapiens is shown in italic directly under the HBD sequence of HBD-EPO.

We developed a synthetic gene encoding for the chimeric HBD-EPO protein consisting of the HBD from D. rerio and human EPO connected with a serine/glycine linker.
The obtained genetic construct was used for creation of bacterial strain for the production of recombinant HBD-EPO in quantities sufficient for performing experiments described below in this article. We also developed the procedure for HBD-EPO purification by column chromatography.
The results of electrophoretic analysis of protein fractions at different stages of HBD-EPO purification are shown in Fig. 2.
Fig. 2. Electrophoresis in 12% polyacrylamide gel of protein preparations obtained during HBD-EPO purification from E. coli M-15 [pREP4, pL610] producer strain: 1, 2) extract of E. coli cells before (1) and after (2) induction of protein synthesis with IPTG; 3) soluble proteins from E. coli cells after the protein synthesis induction with IPTG; 4) washed inclusion bodies; 5, 6) HBD-EPO-containing fractions eluted from WorkBeads 40 S in the sample buffer with (5) and without (6) DTT; 7, 8) HBD-EPO fractions after refolding eluted from heparin-Sepharose CL-6B in the sample buffer with (7) and without (8) DTT; M, protein molecular weight markers, 14-97 kDa (Bio-Rad, USA). The presence (+) or absence (–) of DTT in the sample buffer is shown under the lanes.
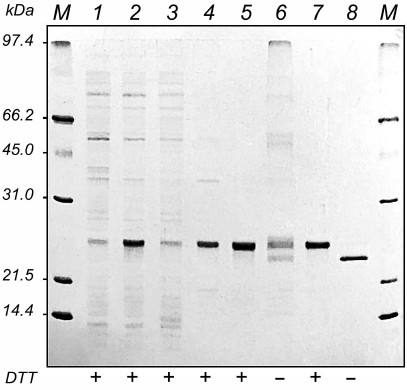
Comparison of electrophoregrams of bacterial cell extracts before and after the protein synthesis induction with IPTG (Fig. 2, lanes 1 and 2) revealed a protein band corresponding to the recombinant HBD-EPO with apparent molecular mass of 24 kDa, which was higher than its calculated molecular mass (20.5 kDa). This protein comprised ~50 % of total cell protein and was synthesized mostly as insoluble inclusion bodies (Fig. 2, lanes 3 and 4). After column chromatography on WorkBeads 40 S, fractions with the maximal absorbance at 280 nm were combined. The total volume of the combined protein fractions was 10 ml; protein concentration was 2.15 mg/ml. Electrophoretic analysis in the presence of DTT showed that the isolated protein was homogenous; however, multiple bands presumably corresponding to differently folded monomeric and oligomeric forms of HBD-EPO were observed in electrophoregrams in the absence of DTT (Fig. 2, lanes 5 and 6).
Protein refolding was performed as described by Wang et al. [16] with slight modifications; HBD-EPO was then subjected to chromatography on heparin-Sepharose CL-6B. The first three eluted fractions (total volume, 20 ml) contained almost homogenous HBD-EPO monomer, as judged by electrophoresis in the presence and absence of DTT. However, in the absence of DTT, the refolded protein had a higher electrophoretic mobility with apparent molecular mass of ~23 kDa (Fig. 2, lanes 7 and 8). Similar increase in the electrophoretic mobility of the refolded protein in the absence of DTT was observed for bacterially produced non-glycosylated 6His-s-tag-EPO [20].
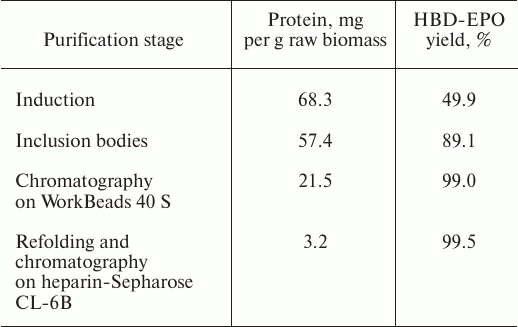
The total amount of purified protein was ~3.2 mg. The data on the HBD-EPO purification efficiency and yield at different purification stages are shown in the table.
Yield of recombinant HBD-EPO at different purification stages
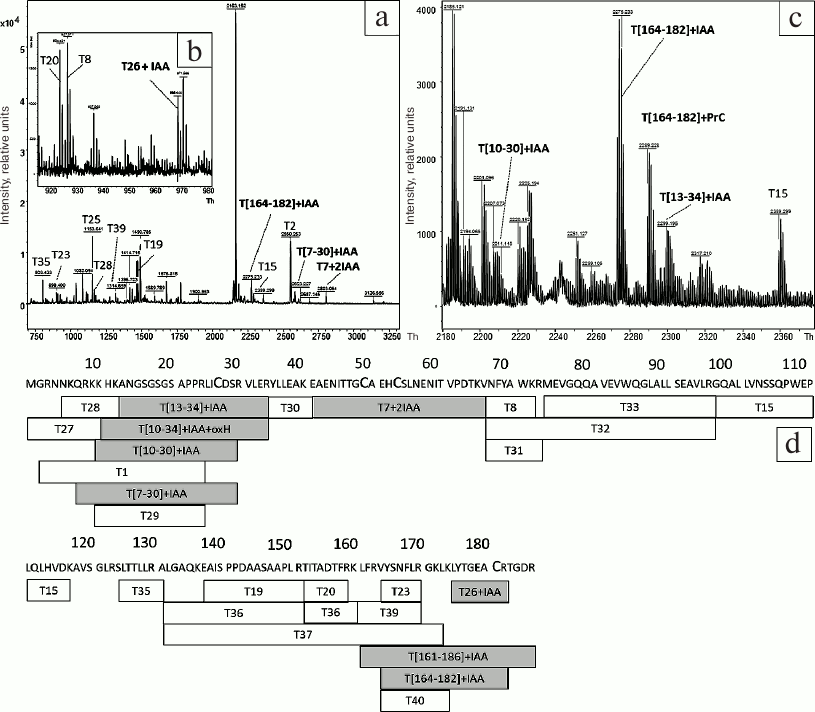
To study the structure of refolded HBD-EPO after chromatography on WorkBeads 40 S, the protein band was cut from the gel (Fig. 2, lane 7) and digested with trypsin with subsequent addition of DTT and to the gel (see “Materials and Methods” for details). The results of MALDI mass spectrometry of the tryptic peptides are shown in Fig. 3, a-c; peptide sequences and their modifications are presented in Table S1 (see Supplement to this paper on the site of the journal http://protein.bio.msu.ru/biokhimiya and Springer site Link.springer.com). Analysis of mass spectra revealed a number of peaks corresponding to the protonated molecular ions of cysteine-containing peptides: T[7-30] + IAA (m/z 2607.3409), T[13-34] + IAA (m/z 2299.1836), T7 + 2IAA (m/z 2803.2123) containing two cysteine residues that form one of the disulfide bridges in the native protein, T[164-182] + IAA (m/z 2275.3167), T[164-186] + IAA (m/z 2704.375), and T26 + IAA (m/z 969.4701). Cysteine residues in these peptides were S-carbamidomethylated. The spectra also had three peaks corresponding to cysteine-containing peptides T[7-30] + IAA + oxH (m/z 2623.3226), T[10-30] + IAA + oxH (m/z 2211.0515), and T[10-34] + IAA + oxH (m/z 2708.3472) containing oxidized histidine residues in addition to iodoacetamide-modified cysteines. The coverage of HBD-EPO sequence with tryptic peptides (Fig. 3d) was 94.6%. The absence of some peptides in the spectra could be explained by their low molecular mass (<700 Da), which was below the detection limit.
Fig. 3. MALDI mass spectra of peptides obtained by tryptic digestion of reduced HBD-EPO (a-c) and diagram showing the coverage of HBD-EPO sequence with tryptic peptides (d). a, b) MALDI mass spectra of tryptic peptides obtained by digestion of the protein band with apparent molecular mass of ~24 kDa (Fig. 2, lane 7) with m/z 700-3300 (a), 910-990 (b), and 2180-2270 Th (c). Before digestion in the gel, the protein was treated with DTT, and cysteine residues were reduced with iodoacetamide. d) Identified peptides are shown in rectangles with peptide name inside (T with peptide number); corresponding amino acid sequences are shown above; IAA, modification with iodoacetamide; oxH, oxidize histidine; gray rectangles, cysteine-containing peptides.
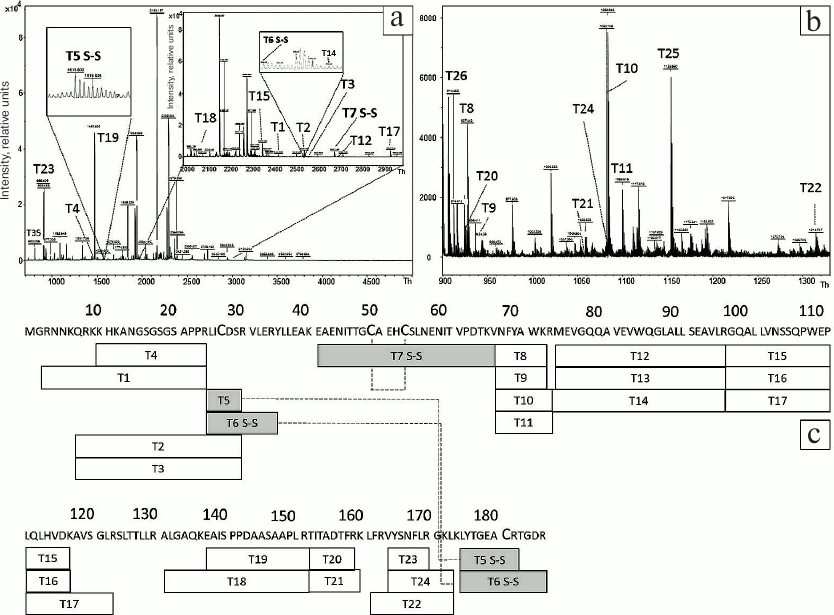
To estimate the efficiency of HBD-EPO refolding, the corresponding protein band was cut out of the gel (Fig. 2, lane 8) and subjected to trypsinolysis in the absence of DTT and iodoacetamide. The samples were loaded on the gel in the DTT-free sample buffer. MALDI mass spectra of the obtained peptides are shown in Fig. 4, a and b; the sequences of these peptides and their modifications are presented in Table S2 (Supplement). The mass spectra lack the peaks corresponding to peptides containing S-carbamidomethyl cysteine (Fig. 3, a and b) but contain peaks confirming the presence of disulfide bonds in the studied protein. Thus, the peak T7 S–S (Fig. 4a) with m/z 2687.1215 confirms the presence of disulfide bond between Cys79 and Cys83. The presence of the second disulfide bridge between Cys57 and Cys211 is corroborated by the peaks T5 S–S and T6 S–S with m/z 1615.6938 and 2542.306, respectively. Therefore, mass spectrometry analysis confirms the presence of two disulfide bridges in refolded HBD-EPO. The absence in mass spectra of peaks corresponding to molecular ions of cysteine-containing peptides with no post-translational modification with iodoacetamide or peptides with modifications such as cysteinyl-S-β-propionamide [30] formed by cysteine reaction with unpolymerized acrylamide monomers indicates that HBD-EPO folded correctly.
Fig. 4. MALDI mass spectra of peptides obtained by tryptic digestion of reduced HBD-EPO (a, b) and diagram showing the coverage of HBD-EPO sequence with tryptic peptides (c). a, b) MALDI mass spectra of tryptic peptides obtained by digestion of the protein band with apparent molecular mass of ~23 kDa (Fig. 2, lane 8) with m/z 970-5000 Th (a) (with two detailed inserts) and m/z 900-1330 Th (b). Before digestion in the gel, the protein was not treated with DTT or iodoacetamide. Dashed lines, disulfide bonds between identified peptides; for other designations, see Fig. 3c.
Activity of purified HBD-EPO was tested in vitro in TF-1 cell line (Fig. 5).
Fig. 5. Dependence of the optical density (measured in the plate well) on the sample protein concentration in the in vitro assay of HBD-EPO (open circles) and Epostim (closed circles, control) activity in TF-1 cells. The data are shown as mean ± standard deviation (n = 3).
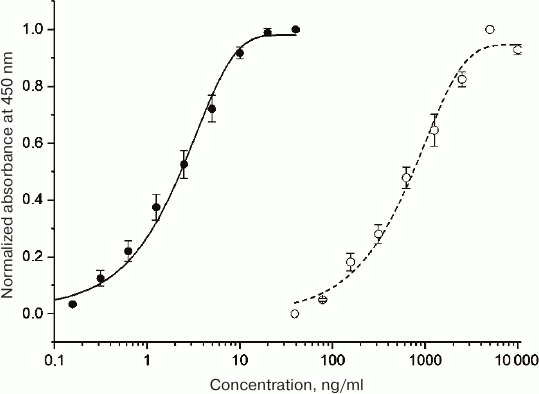
EC50 values for the control epoetin beta produced in eukaryotic cells (Epostim; Farmapark, Russia) and HBD-EPO produced in procaryotes were 2.3 and 614 ng/ml, respectively (average from three plates), i.e., the specific activity of HBD-EPO was less than 0.5% of the specific activity of Epostim and ~4% of the specific activity of bacterially expressed EPO obtained in [20]. Such a low activity of HBD-EPO could be explained by the absence of glycosylation and, probably, by the presence of additional positively charged N-terminal domain (Fig. 1) that can destabilize protein globule or hinder its interaction with cell receptors. We introduced the N-terminal HBD to provide protein retention at the site of bone damage. Despite low activity of recombinant HBD-EPO, we still believed that it would produce required effects upon local application, because an increase in the local HBD-EPO concentration will compensate its low activity, while HBD-EPO immobilization on heparin-containing substrate will promote protein stabilization.
To estimate the principal possibility of HBD-EPO application in experiments on bone tissue regeneration, we had to characterize its activity in vivo. We failed to find published data on the bacterial expression and in vivo characterization of EPO variants, so to estimate the in vivo activity of HBD-EPO, we used the test commonly used for EPO expressed in eukaryotic cells.
Biological activity of HBD-EPO was tested in vivo in 8-week-old CBA mice by counting the number of reticulocytes in blood after injection of the tested preparations using Epostim as a control [26]. Both HBD-EPO and Epostim were injected subcutaneously as equimolar mixtures with heparin (see “Materials and Methods”). The activity of HBD-EPO was 174 IU/mg, which is approximately 100 times lower than the activity of Epostim. Such a low in vivo activity of HBD-EPO might be related to its fast elimination from the circulation. To perform pharmacokinetic studies, we had to develop an adequate system for measuring HBD-EPO concentration in a solution.
The concentrations of HBD-EPO and other EPO variants were measured using commercially available sandwich ELISA kit (Vektor-Best-Evropa, Russia). This kit is advertised as a tool for measuring the concentration of native EPO in patient’s blood serum. However, when we used this kit for determining Epostim concentration, the obtained values were approximately two times lower that the actual values; while the determined concentrations for bacterially expressed HBD-EPO were approximately an order of magnitude lower than the values determined spectrophotometrically.
The ability of commercially available monoclonal antibodies EP1, EP2, EP3, EP5, EP8, and EP25 to interact with HBD-EPO and other recombinant EPO variants [20] was studied by immunoblotting. Out of six antibody clones tested, only EP8 interacted with all bacterially produced recombinant EPOs and Epostim (data not shown).
We also raised immune sera interacting with all EPO variants, both produced in prokaryotes and eukaryotic cells, by immunization of rabbits with purified 6His-MBP-EPO and s-tag-EPO [20], and then chose the serum with the maximal titer (1 : 2500).
Using EP8 antibody and obtained anti-EPO rabbit serum, we developed sandwich ELISA test system for quantitative assay of HBD-EPO and epoetin beta (Epostin) in a solution. Monoclonal EP8 antibody was adsorbed on the plate at a concentration of 1 µg/ml and used for capturing the antigen, while the rabbit serum was used as detecting agent (dilution of 1 : 2000); goat anti-rabbit immunoglobulins conjugated with horseradish peroxidase (IMTEK, Russia) were used at dilution of 1 : 10,000. The developed test system was able to detect EPO proteins in a concentration range from 1 to 100 ng/ml and was used in further experiments.
The results of pharmacokinetic studies in rats are shown in Fig. 6. HBD-EPO was completely eliminated from the rat circulation within 10 h after its subcutaneous injection, while the concentration of Epostim in the samples of blood plasma decreased negligible within 48 h after injection.
Fig. 6. Dependence of the HBD-EPO (diamonds, dashed line) and Epostim (squares, solid line) concentrations in rat blood plasma on the time elapsed from the subcutaneous administration of the protein preparations in a dose of 100 μg/kg body weight. The data are shown as mean ± standard deviation (n = 3).
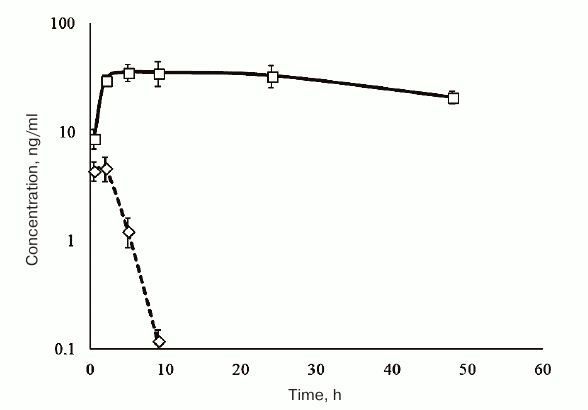
HBD-EPO was injected subcutaneously, because this method of administration increases the elimination half-life of the protein. Indeed, according to the published data, the elimination half-life of Epostim after its intravenous injection in patients is 5-6 h, whereas subcutaneous injection increases it to 16-24 h [31] or even more (as in our experiments).
To study the pharmacokinetics of the tested recombinant proteins and to measure their in vivo activity, we used a mixture of HBD-EPO or Epostim (control) with heparin to screen the positive charge of HBD due to its complex formation with heparin and, probably, to slow down protein release into the blood. The complex of heparin with HBD-EPO is non-covalent (experimentally determined Kd of the BMP-2 complex with heparin is ~20 nM [32]). The concentration of HBD-EPO in its equimolar mixture with heparin used in the experiments was 160 μg/ml, which corresponds to 18 μM, i.e., to the value that is three orders of magnitude higher than the supposed Kd (20 nM). This means that in the injected mixture, all protein was in a complex with heparin. However, considering low rate of protein release to the blood and, therefore, its strong dilution, and heparin redistribution between other heparin-binding proteins, dissociation of the complex in the blood should prevail, so that the protein will be easily eliminated from the organism.
The observed pharmacokinetics of nonglycosylated HBD-EPO and glycosylated Epostim almost completely reproduced the data for EPO variants synthesized in bacterial and eukaryotic cells presented in Wang et al. [16]. Both rh-ng-EPO synthesized in E. coli cells, which has been used in study of Wang et al. [16], and HBD-EPO obtained in our work were almost completely eliminated from the circulation within 10 h, while EPO synthesized in Chinese hamster ovary cells (Sunshine Pharmaceutical Company, China) was found in the blood for longer periods of time, although the rate of its elimination was higher than for Epostim. This means that HBD-EPO combination with heparin for subcutaneous injection does not noticeably increase the elimination half-life of HBD-EPO.
It should be noted that both control and experimental groups of animals displayed the symptoms of impaired hemostasis: blood samples withdrawn after the injection of EPO preparations were very viscous and exhibited signs of hemolysis. In the experimental group, the major signs of hemostasis impairment were observed already at the first hours of the experiment, while in the control group, these symptoms were delayed. We did not perform general and biochemical blood assays; nevertheless, the observed hemostasis impairments suggested pronounced side effects upon systemic administration of both EPO preparations in a dose of 100 μg/kg body weight.
The in vivo and in vitro biological activity of HBD-EPO indicates the functional activity of the obtained protein preparation. We assume that the presence of HBD in HBD-EPO will provide its efficacy upon local administration in small doses due to high local concentrations achieved by HBD-EPO immobilization on DBM. Fast elimination of small amounts of nonglycosylated low-molecular-weight protein through its slow release from DBM into the blood could prevent the development of negative responses that would be induced by large doses of the same preparation.
For further experiments, we developed a method for HBD-EPO immobilization on DBM. DBM disks were incubated with varying amounts of HBD-EPO, washed, and lyophilized as described in “Materials and Methods”. The amounts of non-bound and bound protein were estimated by ELISA. As a result of these experiments, we determined the DBM capacity for the HBD-EPO and s-tag-BMP-2 (at least, 4 mg/g DBM). By using the developed method and by loading the proteins in amounts below the matrix capacity, it was possible to immobilize any required quantity of HBD-EPO, s-tag-BMP-2, or both proteins at a desired ratio.
The quantity of the immobilized proteins and the ratio between them were estimated electrophoretically. Figure 7 shows the results of fractionation in polyacrylamide gel of HBD-EPO, s-tag-BMP-2, and samples eluted from disks by heating them in the sample buffer (see “Materials and Methods”).
Fig. 7. Electrophoresis in 12% polyacrylamide gel of HBD-EPO and s-tag-BMP-2 preparations and proteins eluted from DBM disks: 1) HBD-EPO; 2) s-tag-BMP-2; 3) proteins eluted from DBM disk loaded with 10 μg of HBD-EPO; 4) proteins eluted from DBM disk loaded with 10 μg HBD-EPO and 20 μg BMP-2; M, molecular weight markers.
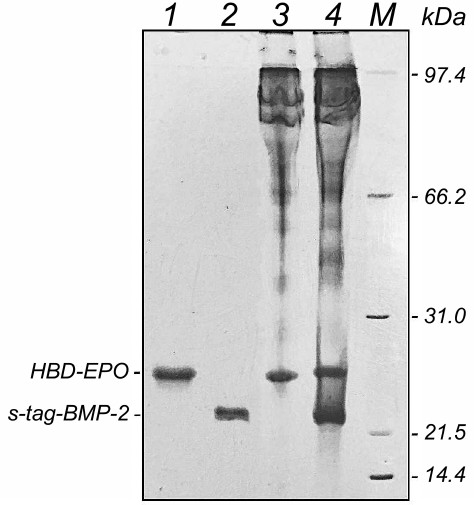
The disks were loaded with 10 µg HBD-EPO or with 10 μg HBD-EPO and 20 μg s-tag-BMP-2. Proteins eluted from the disks (Fig. 5, lanes 3 and 4) had the same electrophoretic mobility as loaded proteins (Fig. 5, lanes 1 and 2). No protein degradation was observed, and the ratio between the proteins (1 : 2) was kept. Lanes 3 and 4 also contained protein bands with higher molecular masses that corresponded to proteins extracted from DBM by the loading buffer as described in “Materials and Methods”.
To verify the possibility of application of DBM disks with immobilized HBD-EPO in experiments on bone regeneration, it was important (i) to elucidate local effects of disk implantation and (ii) to estimate protein release from the disks within the period of observation under in vivo conditions when the disk is washed by bodily fluids.
Two identical DBM disks were implanted subcutaneously in rats into the temporal areas (see “Materials and Methods”) on the opposite sides of the head. Rats of the experimental group received disks loaded with 1 μg of HBD-EPO; the control group received disks loaded with 1 μg of Epostim. After 5 days, the disks were removed. One of the two disks with surrounding tissue fragment was used for histochemical analysis; the second disk was used for measuring residual protein. Disks loaded with 1 μg of HBD-EPO were covered with a connective tissue capsule 1.0-1.5 cm in diameter penetrated with a dense network of blood vessels. Capsule exsection was accompanied with strong bleeding. The disks themselves were of intense red color. Disks loaded with 1 μg of Epostim were covered with less pronounced connective tissue capsule 1 cm in diameter and had lighter coloring. Figure 8 (a, e) shows the disks after their removal on day 5 after implantation and incubation in 6 M guanidine hydrochloride (for extraction of residual protein). The amount of residual protein in the disks loaded with 1 μg of HBD-EPO was 0.9 ± 0.06 μg, as determined with our test system. However, we failed to measure the amount of residual protein in disks loaded with 1 μg of Epostim, which might indicate that Epostim did not bind to DBM and was completely eluted from the disk within 5 days. On the contrary, only 10% of HBD-EPO was eluted from the disks within 5 days after subcutaneous implantation, which means that it was eluted gradually. If the same elution rate preserves (although it usually slows down with time), HBD-EPO will be completely eluted from the disk in 45 days. Such prolonged and gradual elution of factors from the implants is essential for the prolongation of their action and maximal efficacy, in particular, in the bone tissue repair. The maximal activity of BMP-2 requires its slow release from the implant at least for two weeks [33].
Fig. 8. DBM disks removed 5 days after subcutaneous implantation and tissue samples from the implantation areas. a) Disk was loaded with HBD-EPO; b-d) tissue samples after subcutaneous implantation of the disk with HBD-EPO. e) Disk was loaded with Epostim; f-h) tissue samples after subcutaneous implantation of the disk with Epostim. (b, c) and (f, g) General view of tissues from the implantation area (magnification, 25×): 1) dermis; 2) muscle layer (striated cutaneous muscle); 3) adventitia (layer of loose connective tissue); thick white arrows, connective tissue borders. d, h) Peripheral area with vessels (4); magnification, 200×; corresponding areas in panels (c) and (g) are shown with yellow rectangles. b, f) Hematoxylin–eosin staining; c, d, g, h) Heidenhain’s AZAN staining.
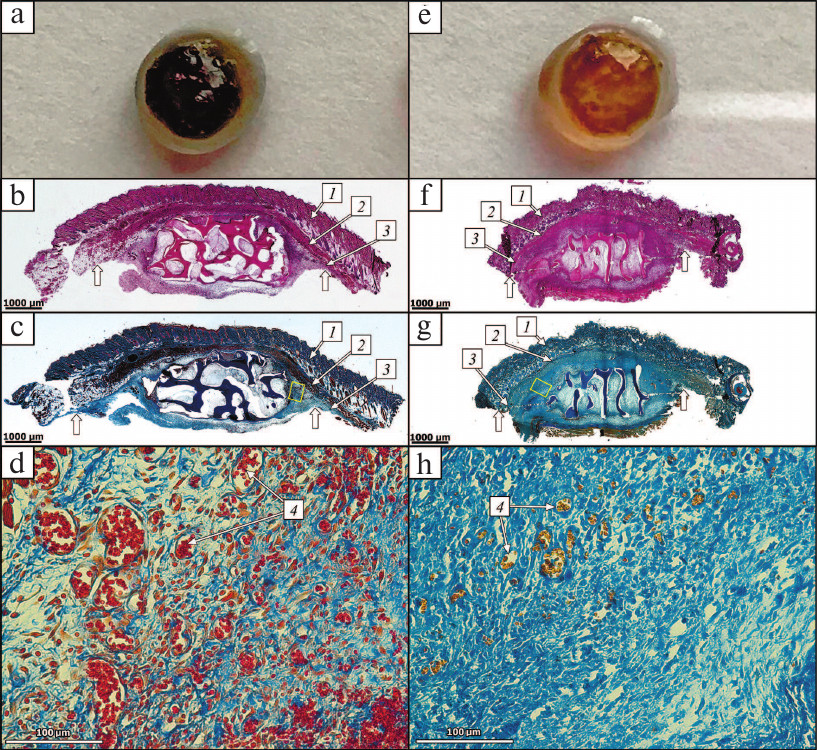
Analysis of histological preparations showed the first stages of connective tissue capsule formation including moderate inflammatory response of the exudative type with the granulation tissue formation. No specific manifestations of cell response to the implant were observed (Fig. 8, b-d, f-h). The general appearance of the preparations was virtually the same in the control and experimental groups: epidermis and its appendages, dermis, hypodermis, and cutaneous muscle were intact; the implant as a fragment of decellularized trabecular bone without signs of its resorption by osteoclasts or perifocal osteogenesis was located in the adventitia; the spaces between the trabeculae contained fibrin and neutrophils. Active inflammation with the formation of immature granulation tissue from fibroblast-like cells and macrophages with moderate number of thin-walled vessels and moderate infiltration of neutrophils including cells with karyorrhexis and karyolysis was observed at the implant periphery. Staining with hematoxylin and eosin (Fig. 8, b and f) showed that preparations from the experimental group were more vascularized that preparations from the control group. Preparations from the experimental group contained areas with well-expressed development of capillary-like thin-walled vessels that showed extravasation of erythrocytes into surrounding connective tissue. To estimate the extent of vascularization, we used Heidenhain’s AZAN staining to identify vessels by contrasting (red or yellowish) erythrocytes [34] (Fig. 8, c, d, g and h) for further histomorphometry (estimation of vessel number and area). The average number of vessels in the periphery of samples from the experimental group (251 ± 27) slightly exceeded the average number of vessels in the control group (204 ± 20). The average vessel diameter in the control group was 15 ± 4 vs. 30 ± 6 μm in the experimental group. The portion of capillaries over 30 μm in diameter in the experimental group was 47%, while in the control group, such capillaries were absent. The average capillary area in the control group was 177 ± 6 vs. 707 ± 10 μm2 in the experimental group (i.e., 4 times higher). The difference in the extent of vascularization in the control and experimental samples is clearly seen in Fig. 8, d and g.
Therefore, HBD-EPO immobilized on DBM disks caused much more pronounced vascularization than Epostim. HBD-EPO administration resulted in a slight increase (by 23%) in the number of vessels with significant (4-fold) elevation in the vessel area in the peripheral zone of the implants. This difference can be explained by the fact that immediately after disk implantation, both preparations induced angiogenesis to the same extent. However, Epostim was completely and rapidly eluted from the implantation area, whereas HBD-EPO was retained on DBM due to presence of HBD in its molecule. HBD-EPO release into surrounding tissues was gradual and lasted for an extended period of time, so that HBD-EPO could stimulate growth of blood vessels. The ability of EPO to cause tissue vascularization by directly acting on endothelial cells is one of its most characterized biological functions [35, 36]. It should be noted that in our experiments, HBD-EPO exhibited angiogenic properties when administered at a sufficiently low dose (1 μg). When used at the same dose, Epostim, which has a significantly higher in vivo activity upon subcutaneous injection but does not bind to DBM, induced local vascularization to a considerably lesser extent than HBD-EPO.
The efficiency of HBD-EPO binding to DBM is similar to that of s-tag-BMP-2 that displays high osteoinductive activity in animal models when immobilized on DBM [22, 37-41]. Besides its action on cell cultures, HBD-EPO induces erythropoiesis and local angiogenesis in vivo. It should be noted that the HBD-EPO in vitro activity (measured in the proliferation tests in human TF-1 erythroleukemia cells) and in vivo activity (estimated from the increase in the reticulocyte count in the blood) observed after HBD-EPO subcutaneous injection were two orders of magnitude lower than for the commercial preparation Epostim. This difference could result from the presence of heparin-binding domain that destabilizes the protein and decreases its ability to bind to the EPO receptor (EPOR). The reduced in vivo activity might be caused by rapid elimination of HBD-EPO from the circulation, which in turn, might result from the lower (~1.5 times) molecular mass of the protein due to the absence of its glycosylation (as compared to Epostim). On the contrary, local angiogenic activity observed after subcutaneous implantation of DBM with immobilized tested proteins was higher for HBD-EPO, which might be related to the retention of HBD-EPO (and not Epostim). This pronounced local angiogenic activity of HBD-EPO gives hope for successful use of this preparation in the development of new highly efficient materials for local application with the purpose of bone regeneration. In our further studies, we plan to characterize osteogenic properties of HBD-EPO and to develop new variants of recombinant EPO with improved in vitro and in vivo activities using the methods of protein engineering by creating different combination of protein domains and replacing domain–domain linkers with more rigid sequences that would reduce negative influence of these domains at each other in the resulting multidomain proteins [42].
Funding
This work was supported by the Russian Science Foundation (project 16-15-00133).
Authors’ Contributions
A. S. Karyagina, A. V. Gromov, and V. G. Lunin proposed research plan and conducted general research management, T. M. Grunina, Z. M. Galushkina, D. A. Tretyak, and I. S. Boksha were engaged in the preparation and purification of proteins, M. S. Poponova and A. V. Demidenko obtained antibodies and performed immunological testing, P. A. Orlova and V. N. Manskikh performed histological studies, N. V. Strukova and M. S. Manukhina worked with laboratory animals, K. E. Nikitin obtained DBM, A. M. Lyaschuk cloned genes, S. A. Cherepushkin tested protein preparations in vitro, N. B. Polyakov, A. I. Solovyev, and V. G. Zhukhovitsky performed mass-spectrometry, A. S. Karyagina wrote this article.
Acknowledgements
We thank D. A. Grumov and A. A. Pereborova for help with mass-spectrometry analysis and discussion of the obtained results, N. A. Lavrova for performing immunoblotting, and L. A. Soboleva for growing bacterial biomass for protein purification.
Conflict of Interests
Authors declare no conflict of interests neither in financial nor in any other area.
Ethical Approval
All procedures with animals were conducted in accordance with the ethical standards of the Gamaleya National Research Center of Epidemiology and Microbiology (Rules for Using Animals in Experiments) and legal requirements adopted by the Russian Federation and international organizations (EU Directive 2010/63/EU and Appendix A of Convention ETS 123).
REFERENCES
1.Krantz, S. B. (1991) Erythropoietin, Blood,
77, 419-434.
2.Shiozawa, Y., Jung, Y., Ziegler, A. M., Pedersen,
E. A., Wang, J., Wang, Z., Song, J., Wang, J., Lee, C. H., Sud, S.,
Pienta, K. J., Krebsbach, P. H., and Taichman, R. S. (2010)
Erythropoietin couples hematopoiesis with bone formation, PLoS
One, 5, e10853.
3.Wu, C., Giaccia, A. J., and Rankin, E. B. (2014)
Osteoblasts: a novel source of erythropoietin, Curr. Osteoporos.
Rep., 4, 428-432.
4.Li, C., Shi, C., Kim, J., Chen, Y., Ni, S., Jiang,
L., Zheng, C., Li, D., Hou, J., Taichman, R. S., and Sun, H. (2015)
Erythropoietin promotes bone formation through EphrinB2/EphB4
signaling, J. Dent. Res., 94, 455-463.
5.Holstein, J. H., Menger, M. D., Scheuer, C., Meier,
C., Culemann, U., Wirbel, R. J., Garcia, P., and Pohlemann, T. (2007)
Erythropoietin (EPO): EPO-receptor signaling improves early
endochondral ossification and mechanical strength in fracture healing,
Life Sci., 80, 893-900.
6.Holstein, J. H., Orth, M., Scheuer, C., Tami, A.,
Becker, S. C., Garcia, P., Histing, T., Morsdorf, P., Klein, M.,
Pohlemann, T., and Menger, M. D. (2011) Erythropoietin stimulates bone
formation, cell proliferation, and angiogenesis in a femoral segmental
defect model in mice, Bone, 49, 1037-1045.
7.Garcia, P., Speidel, V., Scheuer, C., Laschke, M.
W., Holstein, J. H., Histing, T., Pohlemann, T., and Menger, M. D.
(2011) Low dose erythropoietin stimulates bone healing in mice, J.
Orthop. Res., 29, 165-172.
8.Rolfing, J. H. D., Bendtsen, M., Jensen, J.,
Stiehler, M., Foldager, C. B., Hellfritzsch, M. B., and Bunger, C.
(2012) Erythropoietin augments bone formation in a rabbit
posterolateral spinal fusion model, J. Orthop. Res., 30,
1083-1088.
9.Sun, H., Jung, Y., Shiozawa, Y., Taichman, R. S.,
and Krebsbach, P. H. (2012) Erythropoietin modulates the structure of
bone morphogenetic protein 2-engineered cranial bone, Tissue Eng.
Part A, 18, 2095-20105.
10.Rolfing, J. H., Jensen, J., Jensen, J. N., Greve,
A. S., Lysdahl, H., Chen, M., Rejnmark, L., and Bunger, C. (2014) A
single topical dose of erythropoietin applied on a collagen carrier
enhances calvarial bone healing in pigs, Acta Orthop.,
85, 201-209.
11.Patel, J. J., Modes, J. E.,
Flanagan, C. L., and Krebsbach, P. H. (2015) Dual
delivery of EPO and BMP2 from a novel modular
poly-ε-caprolactone construct to increase the bone formation in
prefabricated bone flaps, Tissue Eng. Part C Methods,
21, 889-897.
12.Omlor, G. W., Kleinschmidt, K., Gantz, S.,
Speicher, A., Guehring, T., and Richter, W. (2016) Increased bone
formation in a rabbit long-bone defect model after single local and
single systemic application of erythropoietin, Acta Orthop.,
87, 425-431.
13.Sinyukhin, V. N., Stetsyuk, E. A., and
Lovchinsky, E. V. (1994) Pharmakinetics of human recombinant
erythropoietin, Terapevt. Arkhiv, 66, 60-62.
14.Egrie, J. C., Strickland, T. W., Lane, J., Aoki,
K., Cohen, A. M., Smalling, R., Trail, G., Lin, F. K., Browne, J. K.,
and Hines, D. K. (1986) Characterization and biological effects of
recombinant human erythropoietin, Immunobiology, 172,
213-224.
15.Egrie, J. C., Dwyer, E., Browne, J. K., Hitz, A.,
and Lykos, M. A. (2003) Darbepoetin alfa has a longer circulating
half-life and greater in vivo potency than recombinant human
erythropoietin, Exp. Hematol., 31, 290-299.
16.Wang, Y. J., Liu, Y. D., Chen, J., Hao, S. J.,
Hu, T., Ma, G. H., and Su, Z. G. (2010) Efficient preparation and
PEGylation of recombinant human non-glycosylated erythropoietin
expressed as inclusion body in E. coli, Int. J. Pharm.,
386, 156-164.
17.Jeong, T. H., Son, Y. J., Ryu, H. B., Koo, B. K.,
Jeong, S. M., Hoang, P., Do, B. H., Song, J. A., Chong, S. H.,
Robinson, R. C., and Choe, H. (2014) Soluble expression and partial
purification of recombinant human erythropoietin from E. coli,
Protein Expr. Purif., 95, 211-218.
18.Boissel, J. P., Lee, W. R., Presnell, S. R.,
Cohen, F. E., and Bunn, H. F. (1993) Erythropoietin
structure–function relationships. Mutant proteins that test a
model of tertiary structure, J. Biol. Chem., 268,
5983-5993.
19.Narhi, L. O., Arakawa, T., Aoki, K., Wen, J.,
Elliott, S., Boone, T., and Cheetham, J. (2001) Asn to Lys mutations at
three sites which are N-glycosylated in the mammalian protein decrease
the aggregation of Escherichia coli-derived erythropoietin,
Protein Eng., 14, 135-140.
20.Grunina, T. M., Demidenko, A. V., Lyaschuk, A.
M., Poponova, M. S., Galushkina, Z. M., Soboleva, L. A., Cherepushkin,
S. A., Polyakov, N. B., Grumov, D. A., Solovyev, A. I., Zhukhovitsky,
V. G., Boksha, I. S., Subbotina, M. E., Gromov, A. V., Lunin, V. G.,
and Karyagina, A. S. (2017) Recombinant human erythropoietin with
additional processable protein domains: purification of protein
synthesized in Escherichia coli heterologous expression system,
Biochemistry (Moscow), 82, 1285-1294.
21.Karyagina, A. S., Boksha, I. S., Grunina, T. M.,
Demidenko, A. V., Poponova, M. S., Sergienko, O. V., Lyaschuk, A. M.,
Galushkina, Z. M., Soboleva, L. A., Osidak, E. O., Semikhin, A. S.,
Gromov, A. V., and Lunin, V. G. (2016) Optimization of rhBMP-2
active-form production in a heterologous expression system using
microbiological and molecular genetic approaches, Mol. Genet.
Microbiol. Virol., 31, 208-213.
22.Bartov, M. S., Gromov, A. V., Poponova, M. S.,
Savina, D. M., Nikitin, K. E., Grunina, T. M., Manskikh, V. N., Gra, O.
A., Lunin, V. G., Karyagina, A. S., and Gintsburg, A. L. (2016) Modern
approaches to research of new osteogenic biomaterials on the model of
regeneration of cranial critical-sized defects in rats, Bull. Exp.
Biol. Med., 162, 273-276.
23.Shevchenko, A., Tomas, H., Havlis, J., Olsen, J.
V., and Mann, M. (2006) In-gel digestion for mass spectrometric
characterization of proteins and proteomes, Nat. Protoc.,
1, 2856-2860.
24.Sharapova, N. E., Kotnova, A. P., Galushkina, Z.
M., Lavrova, N. V., Poletaeva, N. N., Tukhvatulin, A. E., Semikhin, A.
S., Gromov, A. V., Soboleva, L. A., Ershova, A. S., Zaitsev, V. V.,
Sergeenko, O. V., Lunin, V. G., and Karyagina, A. S. (2010) Production
of the recombinant human bone morphogenetic protein-2 in Escherichia
coli and testing of its biological activity in vitro and
in vivo, Mol. Biol. (Moscow), 44, 1036-1044.
25.Karyagina, A. S., Boksha, I. S., Grunina, T. M.,
Demidenko, A. V., Poponova, M. S., Sergienko, O. V., Lyashchuk, A. M.,
Galushkina, Z. M., Soboleva, L. A., Osidak, E. O., Bartov, M. S.,
Gromov, A. V., and Lunin, V. G. (2017) Two variants of recombinant
human bone morphogenetic protein 2 (rhBMP-2) with additional protein
domains: synthesis in an Escherichia coli heterologous
expression system, Biochemistry (Moscow), 82,
613-624.
26.Ramos, A. S., Schmidt, C. A., Andrade, S. S.,
Fronza, M., Rafferty, B., and Dalmora, S. L. (2003) Biological
evaluation of recombinant human erythropoietin in pharmaceutical
products, Braz. J. Med. Biol. Res., 36, 1561-1569.
27.Gromov, A. V., Nikitin, K. E., Karpova, T. A.,
Zaitsev, V. V., Sidirova, E. I., Andreeva, E. V., Bartov, M. S.,
Mishina, D. M., Subbotina, M. E., Shevelyagina, N. V., Sergienkov, M.
A., Soboleva, L. A., Kotnova, A. P., Sharapova, N. E., Semikhin, A. S.,
Didenko, L. V., Karyagina, A. S., and Lunin, V. G. (2012) Development
of a procedure for obtaining osteoplastic material based on
demineralized bone matrix with the maximal content of native factor of
bone tissue growth, Biotekhnologiya, 5, 66-75.
28.Osidak, E. O., Osidak, M. S., Sivogrivov, D. E.,
Portnaya, T. S., Grunina, T. M., Galushkina, Z. M., Lunin, V. G.,
Karyagina, A. S., and Domogatskii, S. P. (2014) Kinetics of BMP-2
release from collagen carrier: evaluation by enzyme immunoassay in the
presence of plasma proteins, Bull. Exp. Biol. Med., 158,
104-108.
29.Heidenhain, M. (1905) Zeitschrift fur
wissenschaftliche mikroskopie und fur mikroskopische technik, S.
Hirzel-Leipzig, 22, 339.
30.Xing, G., Zhang, J., Chen, Y., and Zhao, Y.
(2008) Identification of four novel types of in vitro protein
modifications, J. Proteome Res., 7, 4603-4608.
31.Biryukova, L. S., Ushakova, A. I., Kamshilova, N.
I., Ose, I. V., Akimov, A. V., Skobeleva, T. F., and Badmaev, A. L.
(2011) Estimation of safety and therapeutic efficacy of new preparation
stimulating erythropoiesis (epoetin β) in patients subjected to
programmed hemodialysis, Nefrol. Dializ, 13, 128-132.
32.Ruppert, R., Hoffmann, E., and Sebald, W. (1996)
Human bone morphogenetic protein 2 contains a heparin-binding site,
which modifies its biological activity, Eur. J. Biochem.,
237, 295-302.
33.Vo, T. N., Kasper, F. K., and Mikos, A. G. (2012)
Strategies for controlled delivery of growth factors and cells for
bone regeneration, Adv. Drug Deliv. Rev., 64,
1292-1309.
34.Zwingenberger, S., Langanke, R., Vater, C., Lee,
G., Niederlohmann, E., Sensenschmidt, M., Jacobi, A., Bernhardt, R.,
Muders, M., Rammelt, S., Knaack, S., Gelinsky, M., Gunther, K. P.,
Goodman, S. B., and Stiehler, M. (2016) The effect of SDF-1α on
low dose BMP-2 mediated bone regeneration by release from heparinized
mineralized collagen type I matrix scaffolds in a murine critical size
bone defect model, J. Biomed. Mater. Res. A, 104,
2126-2134.
35.Ribatti, D., Presta, M., Vacca, A., Ria, R.,
Giuliani, R., Dell’Era, P., Nico, B., Roncali, L., and Dammacco,
F. (1999) Human erythropoietin induces a pro-angiogenic phenotype in
cultured endothelial cells and stimulates neovascularization in
vivo, Blood, 93, 2627-2636.
36.Kimakova, P., Solar, P., Solarova, Z., Komel, R.,
and Debeljak, N. (2017) Erythropoietin and its angiogenic activity,
Int. J. Mol. Sci., 18, E1519.
37.Gaifullin, N. M., Karyagina, A. S., Gromov, A.
V., Terpilovsky, A. A., Malanin, D. A., Demeshchenko, M. V., and
Novochadov, V. V. (2016) Morphological properties of osteointegration
upon using titanium implants with bioactive coating and recombinant
bone morphogenetic protein rhBMP-2, Morfologiya, 149,
77-84.
38.Andreev, A. Yu., Zakharov, V. D., Zaratyants, O.
V., Borzenok, S. A., Khubenova, M. Kh., Osidak, E. O., Krasheninnikov,
S. V., Karyagina, A. S., and Domogatsky, S. P. (2016) Prospects in the
administration of bone tissue growth factor in the content of collagen
matrix for cornea strengthening (experimental study), Sovr. Tekhnol.
Oftalmol., 4, 11-16.
39.Zakharov, V. D., Andreev, A. Yu., Zaratyants, O.
V., Osidak, E. O., Borzenok, S. A., Krasheninnikov, S. V., Karyagina,
A. S., and Domogatsky, S. P. (2016) Morphological changes in the rabbit
cornea under the action of the bone and cartilage tissue growth factor
rhBMP-2 within the intracorneal collagen implant, Klin. Eksp.
Morfol., 4, 36-42.
40.Zakharov, V. D., Zaratyants, O. V., Andreev, A.
Yu., Osidak, E. O., Borzenok, S. A., Krasheninnikov, S. V., Karyagina,
A. S., and Domogatsky, S. P. (2016) Effect of the growth factor rhBMP-2
in the content of collagen matrix on morphological and biomechanical
properties of retina, Oftalmokhirurgiya, 4, 20-28.
41.Bartov, M. S., Gromov, A. V., Manskih, V. N.,
Makarova, E. B., Rubshtein, A. P., Poponova, M. S., Savina, D. M.,
Savin, K. S., Nikitin, K. E., Grunina, T. M., Boksha, I. S., Orlova, P.
A., Krivozubov, M. S., Subbotina, M. E., Lunin, V. G., Karyagina, A.
S., and Gintsburg, A. L. (2017) Recombinant human Bone Morphogenetic
Protein-2 (rhBMP-2) with additional protein domain synthesized in E.
coli: in vivo osteoinductivity in experimental models on
small and large laboratory animals, Bull. Exp. Biol. Med.,
164, 148-151.
42.Chen, X., Zaro, J. L., and Shen, W. C. (2013)
Fusion protein linkers: property, design and functionality, Adv.
Drug Deliv. Rev., 65, 1357-1369.
Supplementary Tables S1 and S2 (PDF)